



1 Introduction
Various theoretical models and experimental methods have been used to describe field effects on molecular nanoelectronic devices.[1-3] Understanding intra-molecular energy dissipation, transition and conversion processes in field effect electrode-molecule-electrode (E-M-E) systems as molecular thermoelectric devices, has the central role in the development of molecular thermoelectric science, and is important for both prospective applications and fundamental research purposes.[4-15] In addition, considerable attention has been focused recently on the local heat (energy) and charge transfer in single molecule systems, which are the basic building blocks for molecular nanoelectronic systems.[16-22] Understanding of the fundamental mechanisms of heat/charge transport in single molecule nanoelectronic devices as well as the interpretation of the experimental observations requires the development of theoretical methods for the description of open quantum systems at the atomic/molecular scale. Furthermore, study of the local energy transfer in nanostructure semiconductors and organic molecular semiconductor-like systems is motivated industrially toward the miniaturization of thermoelectric devices.[23-25] Thermoelectric characteristics of thermoelectric-like devices vary with size (macro, meso, and nano/molecular), and thus interests for describing heat/electric transport characteristics of these devices have increased during recent years.[26-60] For example, in low temperature regimes, where electron-phonon interaction can be neglected, the thermoelectric figure of merit $(ZT)$, measuring efficiency of a thermoelectric devices, can be given by[15]
$ ZT =\Bigl[ {\frac{GT}{( {\kappa_{{\rm elec}} +\kappa_{{\rm vib(ph)}} } )}} \Bigr] ( {{\bf {\pmb{\textit L}}}^{S}} )^{2}\\ =\Bigl[ {\frac{G}{T( {\kappa_{{\rm elec}} +\kappa_{{\rm vib(ph)}} } )}} \Bigr] ( {{\bf {\pmb{\textit L}}}^{P}} )^{2}\,, $
where $G$ is the electrical conductance, $T$ is the operating/working temperature (the average temperature of the source and drain electrodes), $\kappa_{{\rm elec}}$ is the electronic thermal conductance, $\kappa_{\rm {vib(ph)}}$ is the thermal conductance due to the vibrational degrees of freedom (phonons), ${\bf {\pmb{\textit L}}}^{S}$ and ${\bf {\pmb{\textit L}}}^{P}$ are Seebeck and Peltier coefficients, respectively. The temperature-independence direct-tunneling electric conduction $(G)$ of a single molecule nanoelectronic system can be evaluated using the Landauer formula, i.e. $G\equiv 1/R=( {2e^{2}\tau_{{\rm elec}} (\varepsilon )})/h$, where $h/2e^{2}=12.91~{\rm k\Omega }$ and $\tau_{{\rm elec}} (\varepsilon )$ being the electronic transition function.[48-50] In most field-effect nanoelectronic molecular systems, transmission function $\tau_{{\rm elec}} (\varepsilon )$ represents the electric charge transport (electron transmission) through the molecular system induced by the applied external field.[50] This transmission function, which describes the probability of electron transfer from one contact point to another, can be calculated using Green's function (GF) method based on molecular density of states.\wen{2,23-25} Generally, in the real molecular nanoelectronic circuits, the $\tau_{{\rm elec}} (\varepsilon)$ value and thus molecular electronic conductance depend on the electrode-molecule-electrode coupling constant, $\Gamma$, which is determined by the external bias voltage regime, and molecular contact/junction properties.
$ \tau_{{\rm elec}} (\varepsilon )=\Bigl[ {\frac{1}{4\eta (1-\eta )} +\Bigl( {\frac{E-{E}'}{E_{F} -{E}'}} \Bigr)^{2}\exp (\beta {\kern 1pt}{\kern 1pt}l^{M})} \Bigr]^{-1}\,, $
where $E_{F}$ is the Fermi energy which can be set to zero at low temperatures, ${E}'\approx E_{{\rm HOMO}},l^{M}$ is the potential barrier width, i.e. the effective (field-dependent) length of molecule, and $\beta$ is the tunneling decay length parameter. Also, the asymmetry factor $\eta =\Gamma _{{\rm elec, L(R)}} /\Gamma_{\rm {elec, tot}}~(\Gamma _{\rm {elec, tot}} =\Gamma_{{\rm elec, L}} +\Gamma_{{\rm elec, R}} )$ is a measure of the difference between the left (L) and right (R) electrode-molecule couplings $(\Gamma_{{\rm elec, L}}$ and $\Gamma _{{\rm elec, R}} )$.[51-54] For a symmetric junction, such as ${\rm Au}-{\rm S}-({\rm Ph)}_{\rm n} -{\rm S}-{\rm Au}$ for which $\Gamma _{{\rm elec}} \approx 0.5{\kern 1pt}{\kern 1pt}{\rm eV}$, we have $\eta \approx 1/2$.
Based on quantum coherent tunneling approach, in the low- and high-bias regimes ($\Phi^{\varepsilon } >1~{\rm eV}$ and $\Phi^{\varepsilon } <1~{\rm eV)}$, the tunneling decay length parameter $\beta $ is given respectively by:[55]
$ \beta \approx \beta^{\circ }\equiv 2\alpha \sqrt{\frac{2m_{e}^{\ast } \Phi^{\varepsilon }}{\hslash^{2}}}\quad \text{(at low bias)}\,; \\ \text{and}\;\; \beta \approx \beta^{V}\equiv \beta^{\circ }\sqrt{\Big( {1-\Big(\frac{eV} {2\Phi^{\varepsilon }}\Big)} \Big)}\quad \text{(at high bias)}\,, $
where $\hslash $ is $h/2\pi$, $\Phi^{\varepsilon }$ is the barrier height for tunneling through the LUMO level, $\Phi^{\varepsilon } \approx (E_{F} -E_{{\rm LUMO}} )$, or that of HOMO level, $\Phi^{\varepsilon }\approx (E_{{\rm HOMO}} -E_{F} )$. For most organic molecular nanoelectronic devices, $\Phi ^{\varepsilon }$ is nearly half of HOMO-LUMO gap $({\rm HLG}\equiv E_{{\rm HOMO}} -E_{{\rm LUMO}} )$, and $m_{e}^{\ast } $ is the electron effective mass (here $m_{e}^{\ast } = 0.16 m_{e}$ with $m_{e}$ being the free electron mass). Also, $\alpha \leq 1$ is a parameter that describes the asymmetry in the potential profile across the E-M-E junction; for symmetric cases $\alpha \approx 1$, also in very low temperature regimes, it has been established that non-resonant tunneling is the main conduction mechanism in single molecular devices. In this state, the tunneling probability and decay coefficients of the Landauer formula are given by $\tau_{{\rm elec}} (\varepsilon )\approx \exp (-\beta {\kern 1pt}{\kern 1pt}l^{M})$ and $\beta \approx \beta ^{\circ }$.[48-50]
In low temperature regime, the $\kappa_{{\rm elec}} (T)$ and $\kappa _{{\rm vib(ph)}} (T)$ heat conductions in Eq. (1) are given by
$ \kappa_{{\rm elec}} (T)=\Big( {\frac{2\pi^{2}\tau_{{\rm elec}} (\varepsilon )k_{B}^{2} T}{3h}} \Big)\,, \\ \kappa_{{\rm vib(ph)}} (T)=\frac{k_{B}^{2} T\tau_{{\rm vib(ph)}} (\nu )}{h}\int_{0}^{\nu_{D} } {\frac{x_{k}^{2} {\rm e}^{x_{k} }}{({\rm e}^{x_{k} }-1)^{2}}} {\rm d}x_{k}\,, $
with $x_{k} =\hslash \nu_{k} /k_{B} T$ and $\nu_{D}$ being the Debye cut-off frequency of phonon reservoirs in the gold electrodes $(\nu_{D} \approx 2.16\times 10^{13}~{\rm S}^{-{\rm 1}})$.[58-61] Generally, at very low temperatures $(T\to 0)$, the electronic thermal conductance is approximated by $\kappa_{{\rm elec}} (T)\approx \left( {2\pi^{2}{\kern 1pt}{\kern 1pt}k_{B}^{2} {\kern 1pt}{\kern 1pt}T/3h} \right)\approx \kappa _{{\rm elec}}^{\circ }$ which can be regarded as an electronic quantum universal value of $\tau_{{\rm elec}} (\varepsilon )\approx 1$, approximately.
Also, $\tau_{{\rm vib(ph)}} (\nu )$ in Eq. (4) is the transmission function of the phonons ranging from zero to one at low temperatures. The values of $\tau _{{\rm vib(ph)}} (\nu )$ for the E-M-E devices can be obtained by $\tau_{{\rm vib(ph)}} (\nu )=\Gamma_{{\rm vib(ph)}}^{{\rm tot}} \delta (\nu -\nu_{i} )$ where $\Gamma _{{\rm vib(ph)}}^{{\rm tot}} =(\Gamma_{{\rm vib(ph)}}^{{\rm L}} \Gamma_{{\rm vib(ph)}}^{\rm R} )/(\Gamma_{{\rm vib(ph)}}^{\rm L} +\Gamma_{{\rm vib(ph)}}^{\rm R} )$ with $\Gamma _{{\rm vib(ph)}}$ being the phononic relaxation rate constant.[51] For weakly coupled molecular junctions and quantum dots, we can assume that $\Gamma_{{\rm vib(ph)}}^{\rm L}\approx \Gamma_{{\rm vib(ph)}}^{\rm R}$. In low temperature regimes, the heat current is carried at the molecular junctions mostly by low frequency phonons. In other words, contribution of the high frequency modes to the heat transfer is small because the Debye cut-off of the reservoir spectra is considerably below their frequencies. In addition, at very low temperatures, only the first energy phonon mode $(h\nu_{0})$ contributes significantly to $\tau_{{\rm vib(ph)}} (\nu )$, and thus $\kappa _{{\rm vib(ph)}} (\nu )$ can be simplified to $\kappa_{{\rm vib(ph)}} (T)\approx \left( {\pi^{2}{\kern 1pt}{\kern 1pt}k_{B}^{2} {\kern 1pt}{\kern 1pt}T/3h} \right)\approx \kappa_{{\rm vib(ph)}}^{\circ }$.[28] This quantum of phononic thermal conductance represents the maximum possible value of energy transported per phonon mode at these temperatures (or $\tau_{{\rm vib(ph)}} (\nu )\approx 1$).
In the present work, in continuation and development of our previous works, analogous to the known thermoelectric effects in molecular junction devices, low temperature coefficients of the intra-molecular energy dissipation (${\bf {\pmb{\textit{L}}}}^{{\rm JL}} $, as Joule-like coefficient), energy dissipation/transition ($\bf {\pmb{\textit{L}}}^{{\rm PL}} $, as Peltier-like coefficient), energy conversion ($\bf {\pmb{\textit{L}}}^{{\rm SL}} $, as Seebeck-like coefficient), and energy dissipation/transition/conversion ($\bf{\pmb{\textit{L}}}^{{\rm TL}} $, as Thomson-like coefficient) processes, and the intra-molecular figure of merit $(ZT_{\gamma }^{M})$, are introduced and computed for two candidate molecular nanoelectronic devices. The electronic and vibrational contributions to these intra-molecular thermoelectric-like coefficients (IMTLCs) describing these processes are computed using respectively quantum theory of atoms-in-molecule (QTAIM) and an energy partitioning scheme based on normal modes vibrational analysis.[62-63]
2 Extension of the Concept of Local Heat Distribution to Molecular Devices
In our previous reports,[52-53] we introduced intra-molecular thermoelectric-like effects, intra-molecular thermoelectric-like sections (IMTLS) and intra-molecular thermoelectric-like coefficients (IMTLC) for molecular nanoelectronic devices. In the present work, two intra-molecular partitioning schemes are used for the calculation of the local energy dissipation/transition and IMTLCs. The first includes four intra-molecular sections (IMS), denoted by $S_{\rm I} $, $S_{\rm II} $, $S_{\rm III} $ and $S_{\rm IV} $, as shown in Fig. 1(a), and is used for detailed intra-molecular energy/heat transfer analysis and the local temperature scaling. In the second partitioning scheme, as shown in parts (b), (c) and (d) of Fig. 1, the molecular system is divided into three sets of IMTLSs, either left (L) and right (R), up (U) and down (D), and, or diagonal-left (DL) and diagonal-right (DR) sections, denoted by $(S_{\rm L}$ and $S_{\rm R} )$, $(S_{\rm U} $ and $S_{\rm D} )$ and $(S_{\rm DL} $ and $S_{\rm DR} )$, which are used for the calculation of the local kinetic energy transfer and the corresponding parallel, perpendicular and diagonal intra-molecular thermoelectric-like coefficients (IMTLCs), respectively.

Fig. 1 (Color online) The molecular nanoelectronic device (E-M$_{1}$-E) studied in this work (a), and parallel (b), perpendicular (c), and diagonal (d) intra-molecular partitioning schemes adopted for the analysis of the thermoelectric performance of this system. The Au atoms represent the electrodes. |
To study intra-molecular thermoelectric-like effects, details of the electronic and vibrational responses to the external EF, appearing as charge and energy transfer, should be known. For example, analysis of the EF effect on the local electron density, $\rho (r)$, of the proposed E-M$_{1}$-E molecular system (Fig. 1) is shown in Fig. 2. This E-M$_{1}$-E device can be grouped into $n$-type-like (electron density donor) and $p$-type-like (electron density acceptor) sections. Furthermore, the partitioning (decomposition) of the total molecular energy into a set of additive atomic contributions has been established by the quantum theory of atoms-in-molecules (QTAIM).[64-70] Based on the QTAIM, the atomic electronic energy, $E_{{\rm elec}} (\Omega )$, is given by
$ E_{{\rm elec}} (\Omega )=K_{{\rm elec}} (\Omega )+V_{{\rm elec}} (\Omega )\,, $

Fig. 2 (Color online) Local differential electron density map, $\Delta \rho (S_{i}, \varepsilon )=\sum\limits_{\Omega \in S_{i} }^{N_{S_{i} } } {\rho (\Omega ;\varepsilon )} -{\kern 1pt}\sum\limits_{\Omega \in S_{i} }^{N_{S_{i} } } {\rho (\Omega ;0)} $, obtained for the E-M$_{1}$-E system at EF of $\varepsilon =60\times 10^{-4}~{\rm au}$ strength, determining the donor ($n$-type-like) and acceptor ($p$-type-like) intra-molecular sections, highlighted in red and blue, respectively. |
where $K_{{\rm elec}} (\Omega )$ and $V_{{\rm elec}} (\Omega )$ are total atomic kinetic and potential energies of the atomic basin $\Omega$, respectively. The atomic contributions to the vibrational energies can be expanded as $K_{\rm vib} (\Omega )=\sum\limits_k {K_{\rm vib}^{k} (\Omega )},V_{\rm vib} (\Omega )=\sum\limits_k {V_{\rm vib}^{k} (\Omega )} $ and $E_{\rm vib} (\Omega )=K_{\rm vib} (\Omega )+V_{\rm vib} (\Omega )=\sum\limits_k {E_{\rm vib}^{k} (\Omega )} $, with $K_{\rm vib}^{k} (\Omega ),V_{\rm vib}^{k} (\Omega )$ and $E_{\rm vib}^{k} (\Omega )$ being contributions of the atomic basin $\Omega $ to the kinetic potential and total energies of the $k$-th normal mode. In addition, based on harmonic oscillation (HO) approximation, we found that the potential energies of the $k$-th vibrational normal mode $_{,}V_{\rm vib}^{k} (\Omega )$, forms a portion of $V_{\rm vib}^{k} $ given by[62-63]
$ V_{\rm vib}^{k} (\Omega )\equiv \Biggl[ {\frac{\sum\limits_{q_{\Omega } =x,y,z} {| {l_{k,q_{\Omega } } }|^{2}} }{\sum\limits_{\Omega '} {\sum\limits_{q_{{\Omega }'} =x,y,z} {| {l_{k,q_{\Omega '} } } |^{2}} } }} \Biggr] V_{\rm vib}^{k}\,, $
where $V_{\rm vib}^{k}$ is the potential energies of the vibrational mode $k$ and ${\bf {\pmb{\textit{Q}}}}_{k} =\sum\limits_i {l_{ki} {\bf {\pmb{\textit{q}}}}_{i} } $ is the normal coordinate corresponding to the $k$-th mode defined as linear combination of the 3$N$ mass-weighted nuclear Cartesian coordinates ${\bf {\pmb{\textit{q}}}}_{i} $, with $l_{ki}$ begin their weighting factors. The total energy of atomic basin $\Omega $ is thus obtained by summing its electronic and vibrational contributions as, $E(\Omega )=E_{{\rm elec}} (\Omega )+E_{\rm vib} (\Omega )$.
The intra-molecular inhomogeneous local energy dissipation in a field-effect molecular device due to thermoelectric-like processes arising from different responses of atomic basins of the molecular device to the applied electric field results in an electronic/vibrational temperature-like differences, which can then result in kinetic energy (heat-like) flows across the device. The extents of these heat flows depend on these temperature-like differences. In this scheme, an external EF of strength $\varepsilon $, for each IMTLS $S_{i} $ consisting of $N_{S_{i} } $ atoms induces a change in the local or sectional electronic/vibrational temperature-like difference, $\Delta T_{\gamma } (S_{i},\varepsilon )$, defined by:
$ \Delta T_{\gamma } (S_{i},\varepsilon )=T_{\gamma } (S_{i},\varepsilon )-T_{\gamma } (S_{i},0) \\ \equiv \frac{c_{\gamma } }{k_{B} } \quad\Bigl\{ {\sum\limits_{\Omega \in {\kern 1pt}{S}_{{i}} }^{{N}_{{S}_{{i}} } } {[ {\left\langle {K_{\gamma } } \right\rangle_{\Omega } (\varepsilon )-\left\langle {K_{\gamma } } \right\rangle_{\Omega } (0)} ]} } \Bigr\}\,, $
where $\gamma =$ elec or vib and $c_{{\rm elec}} =2/3$ and $c_{\rm vib} =2$. In this definition, $\left\langle {K_{\gamma } } \right\rangle_{\Omega } (\varepsilon )$ is the electronic/vibrational kinetic energy expectation value of the atomic basin $\Omega $ belonging to the IMTLS $S_{i} $ in the EF strength of $\varepsilon $, and $T_{\gamma } (S_{i},\varepsilon )$ and $T_{\gamma } (S_{i},0)$ respectively denote its electronic/vibrational temperature-like in the presence and absence of EF. In other words, at a given EF strength of $\varepsilon $, the change in the kinetic energy of the IMTLS $S_{i} $ can be regarded as an index of its cooling or warming referenced to its state in the absence of EF $(\varepsilon =0)$. Thus, for each IMTLS $S_{i} $, the intra-molecular sectional heating/cooling due to the electronic/vibrational degrees of freedom $(\gamma )$ can be defined in terms of the variation in its total kinetic energy $K_{\gamma } (S_{i},\varepsilon )=\sum\limits_{\Omega \in {\kern 1pt}S_{i} }^{ N_{ S_{i} } } {\left\langle {K_{\gamma } } \right\rangle_{\Omega } (\varepsilon )}$ as
$ \Delta K_{\gamma } (S_{i},\varepsilon )=K_{\gamma } (S_{i},\varepsilon )-K_{\gamma } (S_{i},0)\,. $
The $\Delta K_{\gamma } (S_{i},\varepsilon )$ value can be considered as the local heating at the $S_{i} $ IMTLS. Based on the values and signs of $\Delta K_{\gamma } (S_{i},\varepsilon )$ for all IMTLSs of the molecule, it can be determined which of them has become electronically and/or vibrationally warmer or colder, and also on a relative scale which of them has become colder or warmer than other IMTLSs at any given EF strength.
3 Phenomenological Analysis of the Intra-Molecular Thermoelectric-Like Effects
In the Onsager phenomenological approach to linear non equilibrium thermodynamics, the general flux of the quantity $\alpha (J_{\alpha } )$ induced by different forces $\beta (X_{\beta } )$ is given by
$ {\bf {\pmb{\textit{J}}}}_{\alpha } =\sum\limits_\beta {{\bf {\pmb{\textit{L}}}}_{\alpha \beta } \cdot {\bf {\pmb{\textit{X}}}}_{\beta } }\,, $
where ${\bf {\pmb{\textit{L}}}}_{\alpha \beta } $ quantities are known as the phenomenological coefficients.[71-78] Starting from this linear relationship, the ${\bf {\pmb{\textit{J}}}}_{Q,\gamma } (S_{i},S_{j} )$ heat flux between the two $(S_{i},S_{j} )$ IMTLSs of a nanoelectronic E-M-E system via the $\gamma $ (electronic or vibrational) degree of freedom can be written in terms of the electric force represented by the electric current $I$, by defining the corresponding intra-molecular thermoelectric-like coefficients (IMTLC) ${\bf {\pmb{\textit{L}}}}_{\gamma } (S_{i},S_{j} )$ as
$ {\bf {\pmb{\textit{J}}}}_{Q,\gamma } (S_{i},S_{j} )={\bf {\pmb{\textit{L}}}}_{\gamma } (S_{i},S_{j} )\cdot I\,. $
Based on the QTAIM analysis presented in the previous section, the ${\bf {\pmb{\textit{J}}}}_{Q,\gamma } (S_{i},S_{j} )$ heat flux can be defined as
$ J_{Q,\gamma } (S_{i},S_{j} ) \approx \frac{Q_{\gamma } (S_{i},S_{j} )}{\Delta t_{\gamma } } \\ \approx \frac{\Delta K_{\gamma } (S_{i},\varepsilon )-\Delta K_{\gamma } (S_{j},\varepsilon )}{\Delta t_{\gamma } }\,, $
where the flown heat $Q_{\gamma } (S_{i},S_{j} )$ is considered to be equal to the differential changes in the kinetic energies of the two $S_{i} $ and $S_{j} $ IMTLSs of the molecule via the $\gamma $ degree of freedom induced by the applied EF of $\varepsilon $ strength, i.e.
$ Q_{\gamma } (S_{i},S_{j} )=\Delta K_{\gamma } (S_{i},\varepsilon )-\Delta K_{\gamma } (S_{j},\varepsilon )\,. $
In other words, the differential changes in the kinetic energies of each IMTLS pair, induced by an EF, can be considered as the kinetic energy (heat-like) transfer between the corresponding pair of IMTLSs, as described pictorially in Fig. 3. This is one of the central ideas of this series of works on the thermoelectric-like effects in molecular nanoelectronic systems.[52] According to the partitioning schemes (Figs. 1 and 2) applicable for each E-M-E system, the $(S_{i},S_{j} )$ IMTLS pair can be $(S_{\text{L}},S_{\text{R}} ),(S_{\text{U}},S_{\text{D}} )$ and/or $(S_{\text{DL}},S_{\text{DR}} )$. The IMTLC ${\bf {\pmb{\textit{L}}}}_{\gamma } (S_{i},S_{j} )$ of each pair of IMTLS can thus be found be rearranging Eq. (10) as:
$ L_{\gamma } (S_{\rm L},S_{\rm R} ) =\frac{{\bf {\pmb{\textit{J}}}}_{Q,\gamma } (S_{\rm L},S_{\rm R} )}{I}\,, \\ {\bf {\pmb{\textit{L}}}}_{\gamma } (S_{\rm U},S_{\rm D} ) =\frac{{\bf {\pmb{\textit{J}}}}_{Q,\gamma } (S_{\rm U},S_{\rm D} )}{I}\,, \\ {\bf {\pmb{\textit{L}}}}_{\gamma } (S_{\rm DL},S_{\rm DR} )=\frac{{\bf {\pmb{\textit{J}}}}_{Q,\gamma } (S_{\rm DL},S_{\rm DR} )}{I}\,, $

Fig. 3 (Color online) Pictorial presentation of the local kinetic energy transfer between different diagonal pairs of IMTLSs, Eq. (12). The intra-molecular/sectional kinetic energy transfer, $\Delta K_{\gamma } (S_{i},\varepsilon )$, and differential kinetic energy between the diagonal pair of sections,$\nabla_{xy, \varepsilon } K_{\gamma }$, and kinetic energy (heat-like) flux between the corresponding pair of IMTLSs, $\Delta_{\gamma } (\varepsilon )/dt_{\gamma } $, in the EF of $\varepsilon $strength, are also shown. |
where $\gamma =$ elec or vib. For simplicity, notations of the intra-molecular thermoelectric-like heat flux ${\rm { J}}_{Q,\gamma } (S_{i},S_{j} )$ and phenomenological coefficient ${\bf {\pmb{\textit{L}}}}_{\gamma } (S_{i},S_{j} )$ will be abbreviated, where needed.
In the present work, based on the linear-response theory, the electronic and vibrational IMTLCs and thus $ZT_{\gamma }^{M}$ are computed based on the QTAIM and HO approximation in low temperature regime, where thermal energy $k_{B} T$ is much smaller than the molecular levels broadening originating from the coupling of the molecule to the leads of the circuit, it can be assumed that the molecular energy levels are time-independent when exposed to a static electric field. Different molecular time scale models have been used to describe electronic and vibrational energy fluxes in nanoelectronic systems. For example, the inverse of the Bohr frequency $(\nu_{B} )$, and the inverse of the Debye frequency $(\nu_{D} )$ are chosen as the electronic and vibrational time scales in E-M-E systems, respectively.[65-66] Therefore, for direct quantum methodology for the evaluation of electronic $({\bf {\pmb{\textit{L}}}}_{{\rm elec}} )$ and vibration $({\bf {\pmb{\textit{L}}}}_{\rm vib} )$ IMETCs, two field-dependent time scale models, attributed respectively to the electronic tunneling time scale $(t_{{\rm elec}})$, and vibration timescale $(t_{\rm vib})$, are used.
The tunneling traverse time $t_{{\rm elec}}$ which takes for the electron to pass across the molecular barrier width $d^{M}$ (which can be approximated by the length of the molecule, i.e. $d^{M}\approx l^{M}$) depends on the potential barrier width ($l^{M})$ and height $(\Phi^{\varepsilon })$ at EF strength of $\varepsilon$.[52,77-78] This traverse time can be defined as
$ t_{{\rm elec}} (\varepsilon )\equiv l^{M}\sqrt{\frac{m_{e} }{2\Phi^{\varepsilon }}}\,, $
in which $m_{e}$ is the mass of electron at rest. The field-induced change in the electronic tunneling time $\Delta t_{{\rm elec}} (\varepsilon )$ is defined as
$ dt_{{\rm elec}} (\varepsilon )\approx \Delta t_{{\rm elec}} (\varepsilon )\equiv t_{{\rm elec}} (\varepsilon )-t_{{\rm elec}} (0)\,, $
with $t_{{\rm elec}} (0)$ and $t_{{\rm elec}} (\varepsilon )$ being tunneling times in the absence and presence of the EF of strength $\varepsilon $, respectively. The $t_{{\rm elec}} (0)$ tunneling time can arbitrary be set to zero. Also, the height of the barrier $\Phi^{\varepsilon }$ can be regarded as the energy spacing between the highest occupied molecular orbital (HOMO) and the Fermi level $E_{F}$, that is $\Phi^{\varepsilon }\approx E_{F} -E_{{\rm HOMO}}$. Furthermore, based on the Franck-Condon principle, electrons move much faster than nuclei, and compared to almost immediate electronic response, the nuclear motion has delayed response to external EF. Therefore, nuclear (vibrational) motion is described by a different time scale model. This vibrational time scale is usually chosen as the inverse of the characteristic frequency, $\nu_{C}$, of the E-M-E system (i.e. $t_{\rm vib} (\nu )\approx 1/\nu_{C} $) which depends on phononic characteristics of the E-M-E devices.[79-80] For the E-M-E systems, the Debye frequency ($\nu_{D} )$ of the gold electrodes $(\nu_{D} \approx (750$--$800)~{\rm cm}^{-{\rm 1}})$ is generally chosen as the upper limit of the characteristic frequency (i.e. $\nu_{C} \leq \nu_{D} $), since the $\nu_{D} $ vibrational mode plays dominant role in the electric-heat transfer in these systems at very low temperatures. The average vibrational time scale, $t_{\rm vib} (\nu,\varepsilon )$, and its change due to the application of electric field, $\left| {\Delta t_{\rm vib} (\nu,\varepsilon )} \right|$, are thus defined as
$ t_{\rm vib} (\nu,\varepsilon ) \equiv \frac{1}{N_{\nu_{D} } } \Bigl[ {\sum\limits_{\nu_{k} <\nu_{D} }{\frac{1}{\nu_{k} }} } \Bigr]\,,\\ \left| {\Delta t_{\rm vib} (\nu,\varepsilon )} \right| \equiv \left| {t_{\rm vib} (\nu,\varepsilon )-t_{\rm vib} (\nu,0)} \right|\,, $
where $t_{\rm vib} (\nu,0)$ and $t_{\rm vib} (\nu,\varepsilon )$ are molecular vibrational time scales evaluated based on the vibrational frequencies of the system respectively in the absence and presence of the EF of $\varepsilon $ strength. Also, $N_{\nu_{D} } $ is the number of frequency modes below the Debye frequency.
4 Modeling Intra-Molecular Energy Dissipation/Transfer in Field-Effect Nanoelectronic Molecular Devices
At low temperatures and under external electric bias, the Joule and Peltier thermoelectric effects play dominant roles respectively in energy dissipation and energy dissipation/transfer processes in nanoelectronic devices. Since, Joule and Peltier heatings are respectively even (symmetrical) and odd (antisymmetric) functions of the external bias,[81-82] it is possible to decompose IMTLCs into two symmetrical and antisymmetrical components, which describe the intra-molecular Joule-like $({\bf {\pmb{\textit{L}}}}_{\gamma }^{\rm JL} )$ and Peltier-like $({\bf {\pmb{\textit{L}}}}_{\gamma }^{\rm PL} )$ coefficients. Thus, we assume that
$ \begin{cases} {\bf {\pmb{\textit{L}}}}_{\gamma, t, q}^{\rm PL} \equiv \dfrac{| {{\pmb{\textit{L}}}_{\gamma, t, q}^{M,f} } |-| {{\pmb{\textit{L}}}_{\gamma, t, q}^{M,r} } |}{2}\,, \\ {\bf {\pmb{\textit{L}}}}_{\gamma, t, q}^{\rm JL} \equiv \dfrac{| {{\pmb{\textit{L}}}_{\gamma, t, q}^{M,f} } |+| {{\pmb{\textit{L}}}_{\gamma, t, q}^{M,r} }|}{2}\,, \\ \end{cases} \,\, q=\begin{cases} x\quad{\rm for }~(S_{\rm L},S_{\rm R} ), \\ y\quad{\rm for }~(S_{\rm U},S_{\rm D} ), \\ xy\quad{\rm for }~(S_{\rm DL},S_{\rm DR} ),\\ \end{cases} \\ \gamma = {\rm elec}~{\rm or}~{\rm vib}\,, $
in which ${\bf{\pmb{\textit{L}}}}_{\gamma, t, q}^{M,f} $ and ${\bf{\pmb{\textit{L}}}}_{\gamma, t, q}^{M,r} $ are the IMETCs for the forward $(f)$ and reverse $(r)$ biases, respectively. Based on the Thomson-Onsager relation $({\bf{\pmb{\textit{L}}}}^{\rm PL}=\bar{{T}}{\kern 1pt}{\kern 1pt}{\bf{\pmb{\textit{L}}}}^{\rm SL})$, the intra-molecular energy conversion coefficient which describes the intra-molecular Seebeck-like effect $({\pmb{\textit{L}}}_{\gamma }^{\rm SL} )$ can be defined as
$ {\bf{\pmb{\textit{L}}}}_{\gamma,t,q}^{\rm SL} \equiv \frac{{\bf{\pmb{\textit{L}}}}_{\gamma,t,q}^{\rm PL} }{\bar{{T}}_{_{\gamma,q} }^{\rm SL} }\,, $
in which intra-molecular temperature model defined in Eq. (7) is adopted for evaluation of the average temperature $\bar{{T}}_{_{\gamma,q} }^{\rm SL} $. In most thermoelectric systems, the Thomson coefficient $({\bf{\pmb{\textit{L}}}}^{T})$ is given by[83]
$ {\bf{\pmb{\textit{L}}}}^{T}=( {{\rm d}{\bf{\pmb{\textit{L}}}}^{P}/{\rm d}T} )-{\bf{\pmb{\textit{L}}}}^{S} =T( {({\rm d}{\bf{\pmb{\textit{L}}}}^{S}/{\rm d}T)} )\,. $
A new formulation and interpretation of the Thomson effect has recently been proposed by Logvinov et al. in which the Thomson heating/cooling coefficient is considered to be directly proportional to Seebeck coefficient even when $({\rm d}{\bf{\pmb{\textit{L}}}}^{S}/{\rm d}T=0)$.[83-84] Results of their study also show that contributions of Thomson effect to temperature changes $(\Delta T^{T})$ in an $n$/$p$-type semiconductor is related to thermal conductivity coefficients $(\kappa_{n},\kappa_{p})$, lengths of the $n$- and $p$-type semiconductors $(l_{n},l_{p} )$, electrode contact cross-sectional area $({A}')$, Seebeck coefficient $({\bf{\pmb{\textit{L}}}}_{n/p}^{S} )$, working temperature $(T_{w})$ and current density $(J=I/{A}')$ via
$ \Delta T^{T} \approx ( {{A}'(l_{n}^{2} l_{p}^{2} ) ({\bf{\pmb{\textit{L}}}}_{n/p}^{S} )^{2}T_{w} J^{2}} )/( {\kappa_{n} l_{n} +\kappa_{p} l_{p} } )^{2}\,. $
Analogously, contributions of intra-molecular Thomson-like effect to temperature changes $(\Delta T_{\gamma }^{\rm TL} )$ of each pair of IMTLS $S_{i}$ and $S_{j} $, as an $n/p$-type-like junction, can be approximated by
$ \Delta T_{\gamma,t,q}^{\rm TL} \approx \frac{{A}' ( {[ {{\bf{\pmb{\textit{L}}}}_{\gamma,t,q}^{\rm SL} } ]^{2}{\kern 1pt}T_{w} J^{2}( {l/2})^{2} })}{\kappa_{\gamma }^{2} }\,, $
in which $l=l_{n} +l_{p},\kappa_{\gamma, S_{i} } \approx \kappa_{\gamma, S_{j} } \approx \kappa_{\gamma } /2$ at low temperatures. Other notations in this equation are defined in Eqs. (4), (18), and (20). According to this model, it is clear that ${\bf{\pmb{\textit{L}}}}_{\gamma }^{\rm TL} $ and thus $Q_{\gamma,q}^{\rm TL} $ are strongly length (size)-dependent. Using Eq. (20), and based on the linear flux-force response theory, ${\rm d}Q^{\rm TL}/t=\kappa \Delta T^{\rm TL}$, and Onsager phenomenological approach to Thomson effect, ${\rm d}Q^{\rm TL}/{\rm d}t\approx ({\bf{\pmb{\textit{L}}}}^{\rm TL} I)\Delta T\approx \kappa \Delta T^{\rm TL}$, for the current density $I$ as the flux and initial temperature gradient between the two IMTLSs $\Delta T$ as the force, the electronic and vibrational intra-molecular Thomson-like coefficient is also given by ${\rm d}Q^{\rm TL} /{\rm d}t=\kappa {\kern 1pt}{\kern 1pt}\Delta T^{\rm TL} \approx ({\bf{\pmb{\textit{L}}}}^{\rm TL} {\kern 1pt}{\kern 1pt}{\kern 1pt}{\kern 1pt}I{\kern 1pt}){\kern 1pt}{\kern 1pt}\Delta T$, and thus
$ {\bf{\pmb{\textit{L}}}}_{\gamma,t,q}^{\rm TL} \approx \frac{\kappa_{\gamma } {\kern 1pt}{\kern 1pt}\Delta T_{\gamma, t, q}^{\rm TL} }{{\kern 1pt}{\kern 1pt}I{\kern 1pt}{\kern 1pt}\Delta T_{\gamma, t, q} }\,. $
Based on above discussion and using Eqs. (15)--(22), the electronic and vibrational intra-molecular Joule-like heating $Q_{\gamma, q}^{\rm JL} $, Peltier-like heating $Q_{\gamma, q}^{\rm PL}$, Thomson-like heating $Q_{\gamma, q}^{\rm TL} $ and their corresponding intra-molecular heat fluxes ${\rm { J}}_{\gamma, t, q}$ can be introduced as
$ J_{\gamma, t, q}^{\rm JL} =\frac{\Delta Q_{\gamma,q}^{\rm JL} }{\Delta t_{\gamma } } \approx [ {{\bf{\pmb{\textit{L}}}}_{\gamma, t, q}^{\rm JL} } ]\;I\,, \\ {\bf{\pmb{\textit{J}}}}_{\gamma, t, q}^{\rm PL} =\frac{\Delta Q_{\gamma,q}^{\rm PL} }{\Delta t_{\gamma } } \approx [ {{\bf{\pmb{\textit{L}}}}_{\gamma, t, q}^{\rm PL} } ]\;I\,, \\ {\bf{\pmb{\textit{J}}}}_{\gamma, t, q}^{\rm TL} =\frac{\Delta Q_{\gamma,q}^{\rm TL} }{\Delta t_{\gamma } } \approx [ {{\bf{\pmb{\textit{L}}}}_{\gamma, t, q}^{\rm TL} \Delta T_{\gamma,t,q}^{\rm TL} } ]\;I\,. $
In addition, based on Eq. (1), the electronic and vibrational intra-molecular thermoelectric-like figure of merit $ZT_{\gamma }^{M} (S_{i},S_{j} )$ at low temperatures, where the phonon-electron interaction can be neglected, is considered to be the sum of the two electronic and vibrational $(\gamma)$ components as
$ ZT_{\gamma, q}^{M} \equiv [ {{\pmb{\textit{L}}}_{\gamma, q, t}^{\rm SL} } ]^{2} \Bigl[ \frac{T{G}}{\kappa_{{\rm elec}} +\kappa_{{\rm vib(ph)}} } \Bigr]\,. $
Finally, it is predicted that in real molecular thermoelectric devices, the $ZT$ increases when $ZT_{{\rm elec}}^{M}$ increases or $ZT_{\rm vib}^{M{\kern 1pt}}$ decreases implying that EF has induced nuclear motion. Therefore, the effective intra-molecular thermoelectric-like figure of merit $ZT_{q,{\rm eff}}^{M}$ in the field-effect molecular devices, can be defined as
$ ZT_{q, {\rm eff}}^{M} \equiv 1-\Bigl[ {\frac{ZT_{{\rm vib},q}^{M} }{ZT_{{\rm elec},q}^{M} }} \Bigr]\,. $
5 Local Intra-Molecular Electronic Energy Dissipation
When external EF is applied on the molecular device, due the coupling between vibrational and electronic degrees of freedom, part of the energy acquired by the molecular device via its electronic degrees of freedom is converted (dissipated) into the nuclear motion, i.e. vibrational energy and thus can be regarded as the unrecoverable (wasted) electronic energy. This means that only the $\Delta E_{{\rm elec}}^{M}$ part of the total differential energy $(\Delta E_{\rm tot}^{M} =\Delta E_{{\rm elec}}^{M} +\Delta E_{\rm vib}^{M} )$ remains available to drive the nanoelectronic thermoelectric device. Thus, the local intra-molecularel ectronic energy dissipation coefficient, $\eta^{dis}(S_{i},\varepsilon )$, can be defined as
$ \eta^{\rm dis}(S_{i},\varepsilon )\equiv \Bigl[ {\frac{\Delta E_{\rm vib} (S_{i},\varepsilon )} {\Delta E_{{\rm elec}} (S_{i},\varepsilon )+\Delta E_{\rm vib} (S_{i},\varepsilon )}} \Bigr]\,, $
where
$$ \Delta E_{\rm elec(vib)} (S_{i},\varepsilon )= \sum\limits_\Omega^{N_{S_{i} } } {E_{\rm elec(vib)} (\Omega,\varepsilon )} \\ \; -\sum\limits_\Omega^{N_{S_{i} } }{E_{\rm elec(vib)}(\Omega,0)}\,. $$
Using Eq. (25), the most and the least energy-dissipative sections (MEDS and LEDS) of the field-effect molecular nanoelectronic systems, having respectively the maximum $\eta _{\max }^{\rm dis} (S_{i},\varepsilon )$ and minimum $\eta_{\min }^{\rm dis} (S_{i} ,\varepsilon )$ values, can then be determined.
6 Computational Details
Geometry optimization and calculation of the structural and electronic properties of the E-M-E devices studied in this work have been carried out at B3LYP/6-31G* level of theory under different EF strengths applied along the $\pm x$ direction (Fig. 1), using G03 program.[85] For the gold atoms of the electrodes, the LANL2DZ pseudopotential is used. The EF effect on the intra-molecular vibrational energy transfer is worked out by following HO vibrational frequencies obtained for the optimized structures. Starting from the electronic wave functions obtained at each EF strength, the atomic basins are determined using density gradient method, and their electronic energies and their changes with EF strength are calculated. All QTAIM calculations are carried out using AIM2000 program package.[86]
7 Results and Discussion
The change in the local charges and energies, induced by applied EF of $\varepsilon $ strength, can be considered respectively as electron density and as kinetic energy (heat) transfer between the atomic basins and IMTLSs. Both of these transport effects may thus contribute to a local temperature change, which is evaluated theoretically by intra-molecular temperature modeling, as defined in Eq. (7), the intra-molecular thermograph derived based on these local temperatures are shown in Fig. 4. In addition, using the QTAIM, the EF effects on the atomic energies and charges of the E-M$_{1}$-E device are studied, and samples of the results are demonstrated in Fig. 5(a).

Fig. 4 (Color online) Intra-molecular atomic electronic (a) and vibrational (b) temperature changes calculated for some representative atoms of the E-M$_{1}$-E system at $\varepsilon =30\times 10^{-4}~{\rm au}$. |

Fig. 5 (Color online) (a) Intra-molecular atomic electronic and vibrational temperature changes of the end gold-sulfur (-Au-S-) junctions (contact points) of the E-M$_{1}$-E system, as functions of the EF strength. (b) The current-voltage $(I$-$V)$ characteristic curve predicted for the E-M$_{1}$-E device. |
Based on Eqs. (2) and (3), and using Ohm's law $(I=V/R)$, the $I$-$V$ characteristic curve of the E-M$_{1}$-E device is approximated at different EF strengths and is shown in Fig. 5(b). This figure shows that the current increases non-linearly with increasing the bias voltage.
Using Eqs. (12)--(21), values of the total local Joule-like $(Q_{{\rm elec},q}^{\rm JL} +Q_{{\rm vib},q}^{\rm JL} )$, Peltier-like $(Q_{{\rm elec},q}^{\rm PL} +Q_{{\rm vib},q}^{\rm PL} )$, and Thomson-like $(Q_{{\rm elec},q}^{\rm TL} +Q_{{\rm vib},q}^{\rm TL} )$ heatings of the E-M$_{1}$-E molecular system shown in Fig. 1, are calculated at different EF strengths and presented in Fig. 6(a). Analysis of these results show that generally $Q_{\rm tot}^{\rm JL} (S_{i},S_{j} )\gg Q_{\rm tot}^{\rm PL} (S_{i},S_{j} )$, and the Joule-Thomson-like heating $(Q_{\gamma,q}^{\rm JL} +Q_{\gamma,q}^{\rm TL})$ plays the dominant role in the local energy dissipation in single molecule nanoelectronic systems. In addition, the calculated values of the local Thomson-like heating, as demonstrated in Figs. 6(b) and 6(c), shows that for both vibrational and electronic degrees of freedom, the value of the diagonal-sectional Thomson-like heating $Q_{\gamma,q}^{\rm TL} (S_{\rm DL},S_{\rm DR} )$ is generally larger than the corresponding intra-molecular parallel $Q_{\gamma,q}^{\rm TL} (S_{\rm L},S_{\rm R} )$ and perpendicular $Q_{\gamma,q}^{\rm TL} (S_{\rm U},S_{\rm D} )$ values because the value and sign of $Q_{\gamma,q}^{\rm TL} (S_{\rm DL},S_{\rm DR} )$ have contributions from energy transfers along both $x$ and $y$ axes.

Fig. 6 (Color online) (a) The total intra-molecular Joule-like, Peltier-like and Thomson-like heat fluxes, ${\rm { J}}_{\rm tot} (S_{\rm DL}, S_{\rm DR} )={\rm { J}}_{{\rm elec}} (S_{\rm DL}, S_{\rm DR} )+{\rm { J}}_{\rm vib} (S_{\rm DL}, S_{\rm DR} )$, calculated at different EF strengths, for diagonal IMTLSs calculated for the E-M$_{1}$-E device. The electronic (b), $Q_{{\rm elec}}^{\rm TL} (S_{i},S_{j} )$, and vibrational (c), $Q_{\rm vib}^{\rm TL} (S_{i}, S_{j} )$, intra-molecular Thomson-like heatings of the parallel, L-R$(S_{\rm L},S_{\rm R} )$, perpendicular, U-D$(S_{\rm U},S_{\rm D} )$, and diagonal, DL-DR$(S_{\rm DL},S_{\rm DR} )$, IMTLSs of the E-M$_{1}$-E device (Fig. 1). All values are normalized to the corresponding largest values set at 100. |
As established and discussed in our previous reports,[62-63] based on the presented model, the IMTLCs can be calculated for any candidate E-M-E molecular device, to effectively design single molecule thermoelectric devices with controlled and known heat-electric interferences. As another example, a small molecular E-M$_{2}$-E device shown in Fig. 7(a) is also studied and its electronic and vibrational IMTLCs for Peltier effect are calculated and plotted respectively in parts (b) and (c) of Fig. 7. These results show that both electronic and vibrational parallel and perpendicular IMPLCs ${\bf {\pmb{\textit{L}}}}_{t,{\rm elec}}^{\rm PL} (S_{\rm L},S_{\rm R})$ and ${\bf{\pmb{\textit{L}}}}_{t,{\rm elec}}^{\rm PL} (S_{\rm U},S_{\rm D} )$, and oscillate with the EF strength. Furthermore, all vibrational IMTLCs, which are not reported here for brevity, such as ${\bf {\pmb{\textit{L}}}}_{t,{\rm vib}}^{\rm PL} (S_{\rm DL},S_{\rm DR} )$, oscillate with EF. Variations in the electric dipole moment $({\rm {\bf \mu }}_{q})$ with EF strength can be regarded as an alternative scalar index of the response of the electric charge distribution in the E-M$_{2}$-E molecular device to electric field.[87-88]

Fig. 7 (Color online) The E-M$_{2}$-E molecular device studied in this work (a), and its electronic (b) and vibrational (c) intra-molecular Peltier-like coefficients (IMPLC) calculated at different electric field strengths. |
Therefore, size of the electric dipole moment vector and its components (in Debye) are calculated for the E-M$_{2}$-E molecular device at various EF strengths. Sample results are demonstrated in Fig. 8(a). Analysis of the calculated values of $\left| {{\rm {\bf \mu }}_{q} } \right|$ and its components shows that electric dipole moment components increases with EF strength almost linearly with low amplitude oscillations, and its variation is similar to that observed for the parallel electronic IMTLCs shown in Fig. 7. Moreover, analysis of the calculated values of the molecular orbital energies, demonstrated in Fig. 8(b), shows that with increasing the EF strength, the HOMO-LUMO gap, ${\rm HLG}(\equiv E_{{\rm HOMO}} -E_{{\rm LUMO}} )$, is decreased and therefore, it is expected that the parallel electronic IMTLCs, and thus parallel $ZT_{{\rm elec}}^{M{\kern 1pt}} (S_{\rm L},S_{\rm R} )$ to be generally increased. This is due to the fact that HLG is a measure of the electron transport barrier in molecular nanoelectronic systems. Smaller HLG value results in higher electric conductivity and less energy dissipation, and thus larger Seebeck IMTLC and figure of merit $ZT_{{\rm elec}}^{M{\kern 1pt}} (S_{\rm L},S_{\rm R} )$. Similar results have been observed for nano-size thermoelectric systems, for which the electronic Seebeck coefficient ${\bf {\pmb{\textit{L}}}}_{{\rm elec}}^{S} $ and electronic thermoelectric figure of merit $ZT_{{\rm elec}} $ are related to the Fermi energy (i.e. $E_{F} \approx (E_{{\rm HOMO}} +E_{{\rm LUMO}} )/2$).[23-25,40]

Fig. 8 (Color online) (a) Size of the electric dipole moment vector and its components (in Debye) at various EF strengths, and (b) electric field effects on the energies of the occupied (O) and virtual (V) molecular orbitals, calculated for the E-M$_{2}$-E molecular device at DFT-B3LYP/6-31G* level of theory using LANL2DZ basis set and pseudopotential for gold atoms. |
It is possible to compare thermoelectric performances of two nanoelectronic systems E-M$_{1}$-E and E-M$_{2}$-E at a given EF strength based on their differential values of ${\kern 1pt}ZT_{q, {\rm eff}}^{M}$ defined in Eq. (23) using relative differential effective intra-molecular thermoelectric-like figure of merit $(\Delta {\kern 1pt}ZT_{q,{\rm eff}}^{M} )$ defined as
$ \quad \%\Delta {\kern 1pt}ZT_{q,{\rm eff}}^{M} \equiv \Bigl[ {\frac{ZT_{q,{\rm eff}}^{E-M_{1} -E} -ZT_{q,{\rm eff}}^{E-M_{2} -E} } {ZT_{q, {\rm eff}}^{E-M_{2} -E} }} \Bigr]\times 100\,, \\ \quad q= \begin{cases} x~{\rm for }~(S_{\rm L},S_{\rm R} ) \,,\\ y~{\rm for }~(S_{\rm U},S_{\rm D} )\,, \\ xy~{\rm for }~(S_{\rm DL},S_{\rm DR} ) \,.\\ \end{cases} $
Using Eq. (26), the $\Delta {\kern 1pt}ZT_{q,{\rm eff}}^{M}$ values are calculated for the E-M$_{1}$-E (Fig. 1) and E-M$_{2}$-E (Fig. 7(a)) molecular devices and are plotted in Fig. 9 as functions of EF strength. These results shows that generally $ZT_{\rm eff}^{E-M_{1} -E} (S_{i},S_{j} )>ZT_{\rm eff}^{^{E-M_{2} -E}} (S_{i},S_{j} )$. Therefore, it can be predicted that the E-M$_{1}$-E device has a higher thermoelectric performance than the E-M$_{2}$-E device when applied in a real nanoelectronic circuit. This higher performance can be attributed to the more extended $\pi$-conjugated system of the E-M$_{1}$-E device compared to that of the E-M$_{2}$-E device. Analysis of the data demonstrated in Fig. 9 shows also that the effective intra-molecular figure of merit ${\kern 1pt}ZT_{q, {\rm eff}}^{M}$ increases non-linearly with EF strength which can be attributed to the non-linear variations of the molecular vibrational characteristics with EF strength.Oscillations in the IMTLCs (and thus thermoelectric performances)with the EF strengthobtained here are compatible with what already observed and reported for the electronic transport coefficients and performances of the ($n$/$p$)-type semiconductors and mesocopic conductors.\wen{23,89-100} By comparing the trends observed in Figs. 7 and 9, it can be said that oscillations of the $ZT^{M{\kern 1pt}}$ and $ZT_{\rm eff}^{M{\kern 1pt}}$ correspond predominantly to the oscillation of the ${\bf {\pmb{\textit{L}}}}_{\rm vib} $ and changes in the local charge distribution with increasing EF strength. Furthermore, it can be predicted that the overall performance $(ZT)$ of a real molecular thermoelectric device increases when $ZT_{{\rm elec}}^M$ increases or $ZT_{\rm vib}^{M}$ decreases.

Fig. 9 (Color online) The relative differential intra-molecular thermoelectric-like figure of merit, $\% \Delta {\kern 1pt}ZT_{q,{\rm eff}}^{M} (S_{i},S_{j} )$, defined in Eq. (26), of the parallel, perpendicular, and diagonal IMTLSs of the E-M$_{1}$-E and E-M$_{2}$-E molecular devices, introduced in Figs. 1(a) and 7(a), respectively. |
Generally, in low temperature regime, we have $\kappa_{{\rm vib(ph)}} \ll\kappa _{{\rm elec}}$. Interestingly, by increasing temperature, it is possible to have equal electronic and phononic heat conductances $\kappa_{{\rm elec}} (T^{\circ })\approx \kappa_{{\rm vib(ph)}} (T^{\circ })$ at a characteristic temperature $T^{\circ}$[87] and thus based on Eq. (23) we have ${\rm Z}T^{M^{\circ }} \approx 1/2( {{\bf{\pmb{\textit{L}}}}^{\rm SL} } )^{2}(l_{L} )^{-1}$, where ${\bf{\pmb{\textit{L}}}}^{\rm SL} \equiv {\bf{\pmb{\textit{L}}}}_{{\rm elec}}^{\rm SL} +{\bf{\pmb{\textit{L}}}}_{\rm vib}^{\rm SL}$ and $l_{L} \equiv \kappa_{{\rm elec}} /GT\approx (\pi^{2}/3)(k_{B} /e)^{2}$ is a Lorenz number (in the Wiedemann-Franz law). Recently, molecular Lorenz number $(l_{L}^{M} )$ of molecular junctions such as isoprene, 1, 3-benzenedithiol and annulene have been studied and it has been shown that the maximum value of $l_{L}^{M,{\rm peak}}$ is given by $L_{L}^{M,{\rm peak}} \approx 7\pi ^{2}/5\left( {k_{B} /e} \right)^{2}$.[88] It can thus be predicted that in an ideal (or good) molecular thermoelectric device, near a very low characteristic temperature $T^{\circ }\ll T_{{\rm room}}$, we have $\sqrt{2{\rm }l_{L}^{M,{\rm peak}} }\ll{\bf{\pmb{\textit{L}}}}_{{\rm elec}}^{\rm SL}$, and thus ${\rm Z}T^{M} \gg1$. Therefore, the factor $\eta \equiv [ {({\bf{\pmb{\textit{L}}}}_{{\rm elec}}^{\rm SL} )/\sqrt{2l_{L}^{M,{\rm peak}} }}]$ can be regarded as a measure of the efficiency of any multi-terminal molecular device at low temperatures. It can also be predicted that by increasing the working temperature $T$ above T$^{\circ}$ where $\kappa_{{\rm elec}} (T)\ll\kappa_{{\rm vib(ph)}} (T)$, the length-dependence of the $ZT^{M} $ is increased because $\kappa _{{\rm vib(ph)}} \propto l^{M}$ at $T^{\circ }\ll T$.[87]
The most and the least energy-dissipative sections, MEDS and LEDS, of the field-effect molecular devices E-M$_{1}$-E and E-M$_{2}$-E, at different EF strengths, are determined based in Eq. (25) and listed in Tables 1 and 2. This local/atomic energy dissipation scaling and its corresponding concepts presented in this work can be applied to extend the thermoelectric analysis down to molecular nanoelectronic systems.
Table 1 The most (least) energy-dissipative section MEDS (LES), $\eta_{\max }^{\rm dis} (S_{i},\varepsilon )(\eta_{\min }^{\rm dis} (S_{i}, \varepsilon ))$ of the proposed molecular nanoelectronic device E-M$_{1}$-E (Fig. 1) induced by external electric field (EF) with various strengths $(\varepsilon)$ applied in the $\pm x$ directions. |
 |

Although the major focus of the present work has been on local energy dissipation/transfer in non-isolated molecular device (E-M-E) system, the methodology developed here can also be used to study local/atomic EF effect, and thus describe IMTLCs in the field effect isolated molecular systems (M). For example, sample results obtained for the isolated molecular system M$_{3}$ (Fig. 10(a)) are presented in Figs. 10 and 11. It should be mentioned here that QTAIM remains still valid for partitioning a molecular system with small violation of the virial theorem.[101-102] The validity of virial theorem for the M3 molecule (introduced in Fig. 10(a)) in the external EF is investigated by calculating the $-V_{{\rm elec}} /K_{{\rm elec}}$ factor. As can be seen from Fig. 10(b), virial theorem is well valid over the whole range of the EF strengths used in this work. This assures validity of the method proposed in this work for the QTAIM evaluation of the atomic responses (Fig. 10(c)) of an isolated molecular system to external EF.
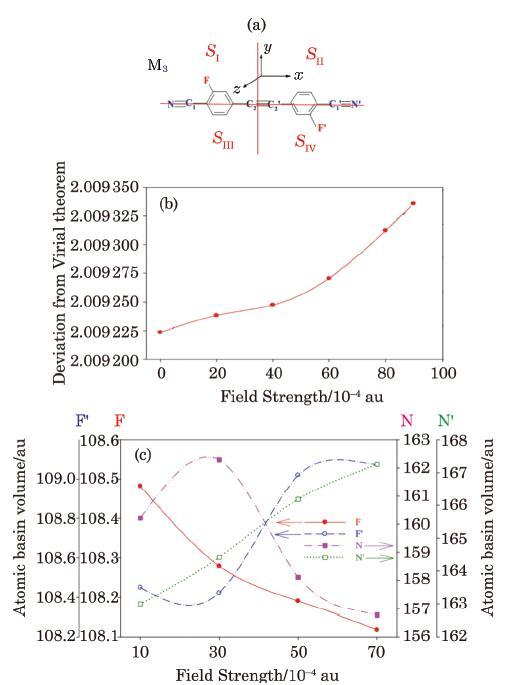
Fig. 10 (Color online) (a) The isolated molecular system M$_{3}$ studied in this work. (b) Examination of the validity of the virial theorem for the M$_{3}$ molecule at different EF strengths. (c) The QTAIM atomic basin volumes as an index of the response of the molecular system M$_{3}$. To external electric field. |
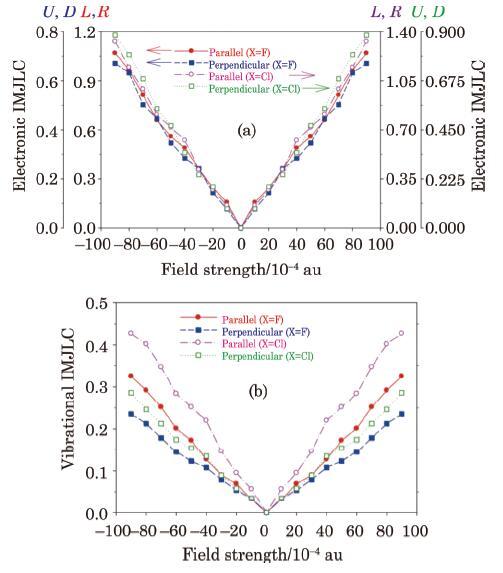
The electronic and vibrational IMJLCs calculated for the M$_{3}$ molecule (Fig. 10(a)) plotted in Fig. 11 shows that the parallel IMJLCs are generally larger than the corresponding perpendicular IMJLCs. In addition, the parallel and perpendicular components of the electronic IMJLCs are larger than corresponding vibrational IMJLCs. The values of the diagonal IMTLCs are nearly similar to what obtained for parallel IMTLCs because of symmetric structure of M$_{3}$.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fig. 11 (Color online) The electric field effect on the electronic (a) and vibrational (b) intra-molecular Joule-like coefficients (IMJLC) calculated for the molecular system M$_{3}$ (introduced in Fig. 11) with two fluorine (X$=$F) and chlorine (X$=$Cl) substitutions. |
Analysis of the results (not reported here for brevity)show that the intra-molecular Peltier-like coefficients (IMPLCs) calculated for the symmetrical molecular system M$_{3}$, are much smaller than the corresponding intra-molecular Joule-like coefficients (IMJLC), and thus for this molecular device, the value of the intra-molecular Peltier-like heating, $Q_{\gamma,q}^{\rm PL} (S_{i},S_{j})$, can be neglected as compared to the value of the intra-molecular Joule-like heating, $Q_{\gamma,q}^{\rm JL} (S_{i},S_{j})$.
Our complementary study showed that the intra-molecular Joule-like and Peltier-like effects occur in both non-symmetrical isolated (M) and non-isolate (E-M-E) molecular systems. Also, analysis of the electrode effects on the IMTLCs shows that numerical values of the electronic IMTLCs for the E-M-E system are generally larger than those of the isolated M system. In addition, based on the results obtained in this study, it is possible to determine which specific atom, group of atoms or sections of a single molecular system are the most appropriate points to be chosen for electrode junctions of interest. Consequently, it is possible to screen proposed E-M-E systems having thermoelectric function of interest with optimum thermoelectric figure of merit.
One advantage of our approach is that it enables us to study substitution (X) effect on the IMTLCs and thus thermoelectric performances. For example, Fig. 11 shows that by replacement of fluorine atoms (X$=$F) with chlorine atoms (X$=$Cl) in the molecular system M$_{3}$, values of the electronic and vibrational IMTLCs are gradually changed with the EF strength, since local/intra-molecular electro-thermal response features depend on the local electronic structures (density and its gradients and Laplacian), bonding and local vibrational characteristics of the molecule and their variation with the EF strength, which are obviously dependent on the type of substitution.
8 Conclusion
A straight forward quantum mechanical methodology is proposed to calculate electronic and vibrational intra-molecular thermoelectric-like coefficients (IMTLC) and intra-molecular thermoelectric-like figure of merits ($ZT_{\gamma }^{M{\kern 1pt}})$ in field-effect nanoelectronic molecular devices usingquantum theory of atoms-in-molecules (QTAIM) and harmonic oscillator (HO) approximation. Introduction of local $ZT_{\gamma }^{M{\kern 1pt}}$ in this work can open a whole new chart of researches to be carried out sequentially to analyze local thermoelectric performancesof any candidate or prospective molecular device prior to its synthesis and/or application in real nanoelectronic circuits. The performance of the real molecular thermoelectric devices $(ZT)$ is improved when $ZT_{{\rm elec}}^{M{\kern 1pt}}$ increases or $ZT_{\rm vib}^{M{\kern 1pt}}$ decreases. Furthermore, the atomic (sectional/local) electronic energy dissipation/transfer scaling presented in this work can be used to effectively design molecular nanoelectronic devices and select the best points for connection to the nanoelectronic circuit of interest by carrying out atomic/sectional response analysis on several proposed molecular nanoelectronic systems with different structures and functional groups acting as connection points or poles and different setups in the nanoelectronic circuit, and determining their efficiencies (local electronic energy transfers) and deficiencies (local energy dissipations).
Although, the IMTLCs (and thus $ZT_{\gamma }^{M{\kern 1pt}}$) introduced in our work cannot reflect all physics of the thermoelectric effects of the working circuit, it can reveal, at least partly, the physical characteristics of the molecular and intra-molecular junctions which have not been accessible so far.
Finally, the model presented in this work may be verified by carrying out comparative theoretical and experimental studies on a couple of well-designed and workable molecular systems having distinctly different figures of merit (obtained from our model) for which actual performances can be measured experimentally by incorporating them in the well-established nano-circuits,[25,89-107] e.g. by probing the characteristics of the junctions and/or sections using AFM/MFM and STM techniques, in addition to the standard current and impedance measurements.
Acknowledgment
Financial supports from the Research and Technology offices of the University of Qom and the University of Isfahan are acknowledged.