



1. Introduction
Proton exchange membrane fuel cells (PEMFCs) are one of the most promising power sources for mobile devices, such as unmanned aerial vehicles and unmanned ground vehicles [1, 2]. The lightweight design brings many challenges to the manufacture of PEMFCs. One of the challenges is the proton exchange membrane material, which should have mechanical flexibility, physical and chemical stability, and most of all, high ion conductivity. In recent years, proton-conducting doped plastic crystals have been proposed to be the candidate as the flexible and anhydrous solid-state proton conductors for PEMFCs [3–10].
Recently, protic organic ionic plastic crystals (POIPCs), which are made up of organic cations and anions via Brønsted acid-base reactions [11–14], have emerged as a subgroup of pure plastic crystals with intrinsic proton conductivity and high thermal stability. Unlike the ordered crystalline structure, the ions in POIPCs are orientationally or rotationally disordered while the centers of mass of ions are aligned in the lattice point (positional order). Thus, POIPCs are less brittle than ordinary crystalline materials and their plasticity helps improve the electrode∣electrolyte contact in PEMFCs [8, 9, 11–13]. Since the cations are Brønsted acid, POIPCs can achieve intrinsic proton conduction without extra proton media (e.g. water). This new type of anhydrous solid-state proton conductor brings many advantages to PEMFCs, including a wider operating temperature range, less catalyst poisoning and simpler design of the fuel cell system [13]. As an organic heterocyclic Brønsted base, the azole-based compounds (e.g. imidazole or 1H-1,2,4-triazole) are often used in POIPCs. The ionic conductivity of imidazolium methanesulfonate ([ImH][CH3SO3]) reaches 1.0 × 10–2 S cm–1 at 458 K in the plastic phase [12]. Furthermore, with a wide plastic phase (360 to 454 K), 1,2,4-triazolium perfluorobutanesulfonate shows a low activation energy for the protonic conduction with a high open circuit voltage (1.05 V) at 423 K (150 °C) for a hydrogen/air fuel cell [13].
According to the experimental results by Forsyth et al the ionic conduction in imidazolium-based POIPCs can be accomplished via either the proton hopping (Grotthuss) mechanism through the crystalline phase or the vehicular motion of the entire cation/anion through a percolated grain boundary phase [14]. For proton conduction in solid-state POIPCs, proton hopping along consecutive hydrogen bond wire is much more efficient since the ions cannot diffuse freely like in the molten state. The early solid-state NMR studies prove that from 240 to 380 K imidazolium ring reorientation process occurs in [ImH][CH3SO3], and this process is often proposed to be the rate-limiting step in proton transportation in imidazole-based materials [15]. The ionic conductivity of [ImH][CH3SO3] rises from the order of 10–4 S cm–1 in Phase II (crystalline phase) to 10–2 S cm–1 in Phase I (plastic crystalline phase) [12].
In the plastic crystal phase, the orientation of the ions is highly disordered, which facilitates the rotational dynamics. As is recently reported by Nazar et al [16–18], the anion rotational dynamics may affect the cation translational motion in inorganic crystalline materials with polyanions through the paddle-wheel mechanism via quasielastic neutron scattering and ab initio molecular dynamics simulations. The high correlation between the rotation of anions and the translational diffusion of the cations prompts us to expand our investigation from alkaline metal cations to the proton diffusion in the POIPC. Mondal et al performed Born–Oppenheimer molecular dynamics (BOMD) simulations on another POIPC, 1,2,4-triazolium perfluorobutanesulfonate, and found that spontaneous auto-dissociation of the N-HN bond in the triazole cation and a complete proton transfer event occurs in a defective crystal with a single proton hole created in the cation [19].
In this article, the local proton hopping process in [ImH][CH3SO3] plastic crystal is investigated via Car–Parrinello molecular dynamics (CPMD) simulation. Inspired by Mondal et al’s work [19], one proton hole was created in the unit cell of [ImH][CH3SO3]. We found that proton hopping occurs directly from imidazolium cation to imidazole molecule, and along with the proton hopping process, the oxygen atom from the anion has a synergetic motion with the transferring proton. The local proton hopping mechanism is expected to provide insights into the material design of azole-based POIPCs and other solid-state azole-based proton conductors.
2. Computational details
The initial crystalline structure is from the single crystal data of [ImH][CH3SO3] at 153 ± 2 K. Though the temperature-dependent powder x-ray diffraction patterns show that at 457 K all the peaks diminish, differential scanning calorimetry measurements show that the plastic crystal phase of [ImH][CH3SO3] remains from 447 to 461 K [12]. To guarantee the model is in the plastic crystal state, we chose 455 K as the targeted simulation temperature. Considering the periodical boundary condition, one unit cell, containing eight pairs of imidazolium cations and methanesulfonate anions, was built to represent the [ImH][CH3SO3] plastic crystal. The simulation cell is an orthorhombic box with the cell parameters of a = 7.918 Å, b = 11.040 Å and c = 16.086 Å. All the simulations were performed via the CPMD method with the CPMD (version 4.1) package [20].
The configuration of [ImH][CH3SO3] unit cell was firstly optimized and then equilibrated at 455 K. A single proton hole was made by removing one hydrogen atom from the imidazolium cation in the optimized configuration. We used this model to mimic the defective crystalline structure in the plastic phase, and the local negative charge will provide a driving force for proton hopping events. The defective crystalline model was first equilibrated for 10 ps, and the sampling run lasted for ∼36 ps. The Nosé–Hoover chain thermostat was used for the whole equilibrium and sampling run [21, 22].
During the whole CPMD simulation, the electronic structure was represented within the generalized gradient approximation to density functional theory using the BLYP exchange-correlation functional, and core electrons were replaced with Troullier–Martins norm-conserving pseudopotentials [23, 24]. The Kohn–Sham orbitals were expanded in a plane-wave basis at the Γ-point up to a kinetic energy cut-off of 80 Ry. The fictitious electron mass in the Car–Parrinello extended Lagrangian was set to μ = 600 a.u., allowing a longer integration time step (5 a.u. ≈ 0.12 fs) for propagating the Car–Parrinello equations of motion [20]. Since the target kinetic energy is not known, we performed a short run without a thermostat to estimate the proper value of the electron kinetic energy. The frequency of the ion thermostat was set to be 6000 a.u. and the frequency of the electron thermostat was set to be 15 000 a.u. to avoid coupling between the ions and electron thermostats. All the nuclei were treated in a classical way, and the nuclear quantum effects of protons are not taken into consideration in this study.
3. Results and discussion
After an equilibrium run at 455 K, the orientation of ions in plastic crystal phase has deviated from the ordered configuration at a lower temperature. The configuration in the defective crystal is different from that in the original crystalline structure. In the original structure of [ImH][CH3SO3], the ions form one-dimensional chain with the N–H…O hydrogen bonds between the imidazolium cations and the methanesulfonate anions. In the defective model, this consecutive hydrogen bond chain breaks at the proton hole. The neutral imidazole molecule forms a hydrogen bond with the nearest imidazolium cation of the other hydrogen bond chain. It is seen in the snapshot (figure 1) that near the proton hole one imidazolium cation rotates at a small angle and forms a hydrogen bond with the neutral imidazole molecule. The neighboring anion also rotates at a small angle, with one oxygen atom pointing to the proton hole. In this local configuration, we labelled the donor and acceptor nitrogen atoms with D and A, and the proton involved in the hydrogen bond as H. The two-dimensional distribution of P(rAD, ∠HDA) (figure 2(a)) shows that there is strong hydrogen bonding interaction between the imidazolium cation and the neutral imidazole. The peak of rAD falls in the range of around 2.75 Å, which is shorter than the average rAD between neutral imidazole molecules in a liquid state (2.9 Å) [25]. The peak of ∠HDA is around 10°, indicating that the hydrogen bond of D–H…A is almost linear, which is just slightly greater than that in ImH/Im mixed solution (8.5°) [26]. According to the theoretical computation, when rAD is less than 2.74 Å, the energy barrier for proton transfer between the imidazolium-imidazole dimer is less than 3.3 kcal mol−1[27, 28]. It suggests that at high temperatures, such as the temperature range of the plastic crystal phase, the energy barrier of proton hopping in between imidazolium-imidazole dimers can be overcome via thermal fluctuation.
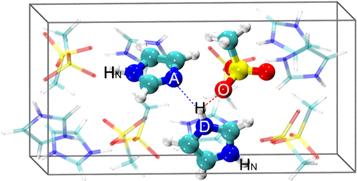
Figure 1. The snapshots of equilibrated [ImH][CH3SO3] unit cell model with one proton hole at 455 K. Color scheme: N, blue; C, cyan; S, yellow; O, red; H, white. A hydrogen bond (blue dashed line) forms between the neutral imidazole molecule and one imidazolium cation. The imidazolium-imidazole dimer, together with the adjacent methanesulfonate anion, is shown in CPK mode. The labeled atoms, D, A and H, represent the donor, the acceptor and the transferring proton, respectively. The labeled O is the nearest oxygen atom in the sulfonate group of the anion. The non-transferring hydrogen atoms are labeled as HN. |

Figure 2. (a) The two-dimensional distribution function P(rAD,∠HDA); (b) the site–site distribution among the imidazolium-imidazole dimer and the adjacent anion. The site label is marked in figure 1. |
The distance distribution in figure 2(b) indicates that proton hopping events do occur during the whole trajectory. The first peak of both rAH and rDH is at ∼1.10 Å, which is the typical N–H bond length. Also, the first peak of rAH is much higher than that of rDH, and the second peak of rDH is ∼1.7 Å. Since the identities of marked atoms (A, D, H and O) do not change during the whole simulation, it can be inferred that the proton hops between the A and D sites. We checked the trajectory movie and found that proton hopping events only occur within the imidazolium-imidazole dimers, while neither the long-range migration of the proton nor the translational diffusion of ions happens during the whole simulation. This local proton hopping behavior is also reported in the 1,2,4-triazolium perfluorobutanesulfonate plastic crystal by Mondal et al using BOMD simulation [19]. Besides, the much higher intensity of the first peak of rAH than rDH indicates that once the proton bond with the accepter nitrogen, it hardly transfers back to the donor nitrogen. Such an observation is in agreement with the BOMD simulation by Mondal et al [19], who found that the proton can bond to the acceptor nitrogen atom for more than 4 ps, during the 5 ps BOMD simulation. The reason for the absence of long-range migration of the proton may be due to the limited size of the simulation cell, which means the negative charge of the proton hole is periodic and the electrostatic repulsion prevents proton migration in one direction. On the other hand, the reorientation of imidazole rings is relevant to the proton transfer process.
The ring reorientation is investigated by time auto-correlation function C(t) of a ring normal vector, which is defined as
$\begin{eqnarray}C\left(t\right)=\left\langle \mathop{{R}_{i}}\limits^{\longrightarrow}\left(t\right)\,\cdot \,\mathop{{R}_{i}}\limits^{\longrightarrow}\left(0\right)\right\rangle ,\end{eqnarray}$
where $\mathop{{R}_{i}}\limits^{\longrightarrow}\left(t\right)$ and $\mathop{{R}_{i}}\limits^{\longrightarrow}\left(0\right)$ is the normal vector at time t and t = 0. Figure 3 shows the decays of the imidazole ring with the labels D and A, and the other six imidazolium rings. All the three curves decay fast within ∼1 ps, and large fluctuation appears after a few picoseconds because of the short simulation time. The fast decay highlights reorientation of the imidazole ring is active in the plastic crystal phase, which is favorable for proton transfer within the imidazolium-imidazole dimer. Among the three curves, the decay of D is slower than the other two. It indicates that the reorientation behaviour of A is similar to the other six imidazolium rings and the transferring proton may attach to site A to form one imidazolium cation.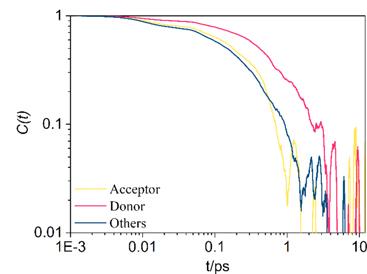
Figure 3. Time correlation functions of the normal vectors of the imidazole ring plane. The acceptor and the donor refer to the imidazole ring marked with A and D, respectively. The others are the other six imidazolium cations not involved in the proton transfer process. |
Besides the hydrogen bond formed within the imidazolium-imidazole dimer, the transferring proton also interacts with the negatively charged O site in the anion. For a clear scenario of the interaction with the oxygen atom, the time evolution of site–site distances is averaged over all the proton hopping events in the trajectory. The time t = 0 ps is defined as the formation of the Zundel-like complex, which is characterized by the reaction coordinate δ (δ = rAH−rDH). The criterion of the Zundel-like complex is ∣δ∣ < 0.1 Å. With this criterion, 28 proton hopping events are counted during the whole trajectory.
The time evolution of rAD and rOH before and after proton hopping is illustrated in figure 4. When the Zundel-like complex is formed at t = 0 ps, the rAD has a minimum of ∼2.59 Å, while the rOH exhibits a maximum of ∼2.85 Å. Before and after the Zundel-like complex, the changing trend of rAD and rOH is opposite on the whole. Therefore, the rAD and rOH may have a negative correlation. The correlation of rAD and rOH can be estimated by the P(rAD, rOH) distribution in the four quadrants that are split by the two means of rAD and rOH. Among all the points, 71% points are in the second and the fourth quadrants, indicating that rAD and rOH have a negative correlation. The covariance value M is estimated by the following function
$\begin{eqnarray}M=\frac{1}{N}\displaystyle \sum _{i=1}^{N}\left({\mathrm{AD}}_{i}-{E}_{\mathrm{AD}}\right)\left({\mathrm{OH}}_{i}-{E}_{\mathrm{OH}}\right),\end{eqnarray}$
where N is the sampling number (29 598 in this work); ADi and OHi are the instant values of rAD and rOH in every simulation step, respectively; EAD and EOH are the mean values of rAD and rOH, respectively (1.96 Å and 2.51 Å in this work, respectively). The covariance value is −0.05 calculated by the function, indicating again that there is a negative correlation between rAD and rOH.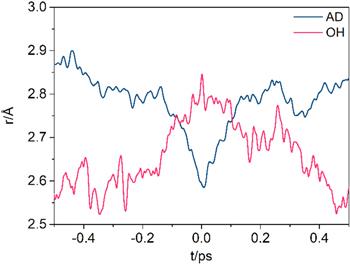
Figure 4. Time evolution of the site–site distances before and after the proton hopping events. The zero point, t = 0 ps, corresponds to the Zundel-like complex. The criterion of the Zundel-like complex is ∣δ∣ < 0.1 Å where δ is the reaction coordinate. |
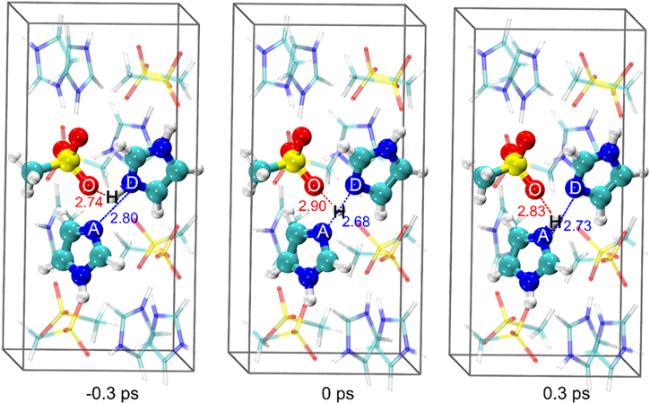
The snapshots of the proton hopping process between A and D are presented in figure 5. At t = 0 ps, the transferring proton is shared by A and D with rAD = 2.68 Å, and rOH is 2.90 Å. When t = −0.3 ps rDH exhibits a standard N−H bond length (1.10 Å) rOH = 2.74 Å and rAD = 2.80 Å. In contrast, when t = 0.3 ps the H atom is bonded with A, in which rAH also indicates a standard N−H bond length (1.10 Å) and in the meantime rOH = 2.83 Å and rAD = 2.73 Å.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. The snapshots of a proton hopping process. The instant values of rOH (in red) and rAD (in blue) are listed in Angstrom (Å). |
4. Conclusions
The local dynamics of [ImH][CH3SO3] in Phase I (plastic crystal phase) are investigated via CPMD simulation. Throughout the whole simulation, both the cations and the anions show local disorders. A proton hole is produced to detect the proton transfer process. We found that proton shuttling events occur between two neighbouring imidazolium cations, and along with proton shuttling there is a synergetic motion of the oxygen atoms in the methanesulfonate group. This behaviour indicates that in the plastic crystalline phase there is structural disorder of imidazolium rings, and the aid of anion can facilitate Grotthuss-type proton hopping in imidazolium-based POIPCs which provides insights into the material design of azole-based POIPCs and other solid-state azole-based proton conductors.