



1. Introduction and motivation
A new class of materials called diluted magnetic semiconductors (DMSs) is obtained by injecting spins into nonmagnetic semiconductors. That is to say, a nonmagnetic semiconductor doped with a very small amount of transition-metal impurities may be endowed with some interesting magnetic properties. It is true that the electronic charge was the only parameter considered in classical semiconductors, however, in DMS materials, the spin of the electron has been found to be an additional degree of freedom that must be considered. This second degree of freedom is needed to comprehend and predict the behaviors of these materials. This was the original idea that allowed an accurate description of the giant magnetoresistance (GMR), independently discovered by A. Fert and P. Grünberg in 1988. This new discovery has allowed the manufacture of novel devices for spintronics and information processing. DMS compounds have garnered attention and have the potential to be very attractive in the fields of spintronic and/or photovoltaic applications [1–5] for many possible reasons. With this new category of materials, greater miniaturization can be achieved, since the spin interaction is smaller than the Coulomb interaction. Furthermore, electric current can flow easily; thus, energy consumption, which is the bottleneck of electronic devices, is reduced considerably. Moreover, DMS compounds offer new functionalities; for instance, the classical bits (zero and one) can be replaced by quantum bits (qubits), creating an opportunity for quantum computing. DMS materials include various systems and a complete review of all their categories is not the goal of this article. Hence, we will restrict ourselves to mentioning the most commonly studied systems. In this respect, several experimental and theoretical studies have investigated the physical properties of DMS materials [6–10]. More specifically, in the spintronic area, various undoped compounds and compounds doped and co-doped with metallic elements have been experimentally and theoretically investigated, such as CuS [11], GaN [12], PbS [13], ZnO [14, 15], and ZnS [16]. The primary property that should be associated with all DMS materials designed for spintronic applications is half-metallicity. This feature exhibits an electronic spin polarization near the Fermi level [17]. Investigations have been performed to develop new systems endowed with this property at ambient temperatures [18, 19]. Exhaustive theoretical studies have been conducted to predict half-metallicity and the value of the Curie temperature TC [20].
This paper describes density functional theory (DFT) calculations carried out for rutile germanium oxide, a semiconductor. Our objective is to calculate (using the local density approximation (LDA) and the LDA combined with the self-interaction correction approximation (LDA-SIC)) the band structure and the density of states (DOS) with the objective of predicting the spin polarization near the Fermi level and the critical temperature of this material. These calculations will demonstrate whether a GeO2 semiconductor with a rutile crystallographic structure may or may not be considered to be a DMS material. It should be underlined that even if the results are encouraging, the critical temperature should be greater than the ambient temperature for any eventual spintronic applications. The expected magnetic behavior in this semiconductor will be studied for rutile GeO2 doped and then co-doped with Co and Fe transition metals. The doping concentration is set to 10%. In fact, this value remains smaller than the percolation threshold (16.66%) to avoid any phase transition of the system leading to crystallographic structure change. The choice of rutile germanium oxide semiconductor is due to its thermodynamic stability under ambient conditions in contrast with that of the α-quartz GeO2 polymorph [21]. Several studies, both experimental and theoretical, have been devoted to the study of the optoelectronic and piezoelectric properties of GeO2 compounds [22–28]. In addition, this material is known for its higher electron mobility and lower operating voltage [29]. Some recent works have also shown that this compound is not only a great candidate for the lithium battery industry [30], but also as a suspended material in fluids to improve their thermal performance [31]. This modern type of heat transfer fluid (nanofluid) has received special attention thanks to its extraordinary physicochemical properties, which are found to be absent from conventional fluids [32, 33]. However, to the best of our knowledge, no theoretical research into the half-metallicity of rutile GeO2 has been reported in the research literature to date. In summary, to examine the possibility of a new material for the spintronic domain, we decided to carry out theoretical calculations for a rutile germanium oxide semiconductor. The results obtained provide valuable evidence of half-metallicity, and this research may contribute to the incorporation of rutile GeO2 in future electronic devices.
2. Computational details and rutile GeO2 structural parameters
2.1. KKR code and the LDA and LDA-SIC approximations
We used the ab-initio calculation method based on DFT and the Akai-KKR (Machikaneyama 2002) code [34] to investigate the effects of magnetic impurity (Co and Fe) doping and co-doping on the behavior of a rutile GeO2 semiconductor. This method has given results that converge with those experimentally obtained [35–40]. Here, the doping elements will replace germanium ions in the crystal structure of the semiconductor. This study is realized to predict the possible usefulness of this material as a DMS material in the areas of spintronics and optoelectronics. To achieve our purpose, we calculated the band structure to evaluate the nature and value of the bandgap. The total and partial DOS were plotted using two approaches, i.e. the LDA and LDA-SIC approximations. These plots allowed us to state that magnetic behavior appears in our doped material and that spin polarization is observable in all cases. The critical temperature TC is also a key parameter to be taken into consideration since TC should be higher than the ambient temperature for any practical DMS in the spintronic field. For this reason, the calculation of this physical quantity is of great importance.
The SIC approximation was used within muffin-tin spheres as the basis set in the KKR-CPA code. In addition, the scalar relativistic approximation was implemented to account for the relativistic effect. It should be noticed that the potential form is provided in an approximate manner by the muffin-tin model. In this model, the wavefunctions in the muffin-tin spheres are developed for the real harmonics up to l = 2, where l is the angular magnetic momentum defined at different sites. In this regard, higher K-points up to 500 were used in the irreducible part of the first Brillouin zone.
2.2. Crystal structure and the parameters of rutile GeO2
At ambient temperature and pressure, three stable polytypes of germanium oxide (GeO2) can exist [41]: an α-quartz-like (P3221) hexagonal structure [42], a rutile-like tetragonal structure with a P42/mnm space group [43], and an amorphous glassy structure [44]. Rutile GeO2 is thermodynamically stable at room temperature [45, 46]. For this reason, this work focuses on investigating the rutile crystal structure. The rutile GeO2 crystallographic structure is chemically and structurally similar to those of other materials, such as SnO2, SiO2, and TiO2 [41].
Rutile belongs to the tetragonal Bravais lattice group with the lattice parameters a = b ≠ c and α = β = γ = 90°. The experimentally reported lattice constants are a = b =4.4066 Å, while c = 2.8619 Å [47]. The unit cell represented in figure 1 below contains two types of atom: Ge in the positions (0.4513 ± 0.0001, 0, 0), whereas oxygen atoms (O) occupy the positions (0.3969 ± 0.0005, 0.3021 ± 0.0005, 0.0909 ± 0.0005) [47, 48]. In the Wyckoff convention (1948), these positions are (3a) and (6a) for Ge and O, respectively. Figure 1 was plotted using the Vesta software (Visualization for Electronic and Structural Analysis) [49].
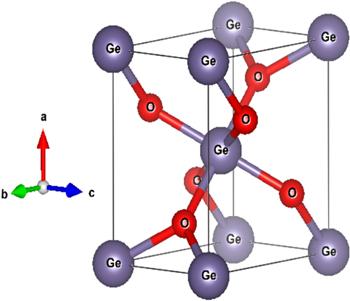
Figure 1. Schematic of the unit cell of the rutile structure of GeO2; the large spheres (grey) represent Ge, while the small spheres (red) represent O. |
Previous theoretical and experimental studies were performed to determine the lattice parameters of the rutile GeO2 structure. Table 1 presents the results of some of these investigations.
Table 1. Parameters of rutile GeO2 according to selected research. |
3. Results and discussion
3.1. Band structure
In figure 2, we have plotted the calculated band structures of pure rutile GeO2 along with the peak symmetry points in the Brillouin zone using the LDA approximation (top figure) and the LDA-SIC approximation (bottom figure). For both approximations, these plots reveal the existence of a direct bandgap in the Γ wavevector, and the positions of the Fermi levels demonstrate that rutile GeO2 is an N-type semiconductor. In the case of the LDA approximation, the value of the bandgap is about 2.10 eV, while an approximate value of 4.80 eV is obtained using the LDA-SIC approximation, which means that this semiconductor has an ultrawide bandgap. These results are consistent with the fact that the LDA approximation underestimates the bandgap value in strongly correlated systems since it does not consider the correlation energy of electrons, as mentioned earlier. The values obtained using both approximations are compared with some of the results published in the research literature and presented in table 2. It can be seen that in our case, the value predicted by LDA is smaller than the experimental value (4.68 ± 0.002 eV) measured using the UV adsorption technique, while our values calculated using the LDA-SIC approximation are slightly greater than this experimental result. In contrast with the theoretical value calculated using the HSE06 hybrid functional (4.64 eV) method, our value calculated using the LDA is smaller; however, the one calculated using the LDA-SIC approximation is almost equal to this theoretical value [54, 55].
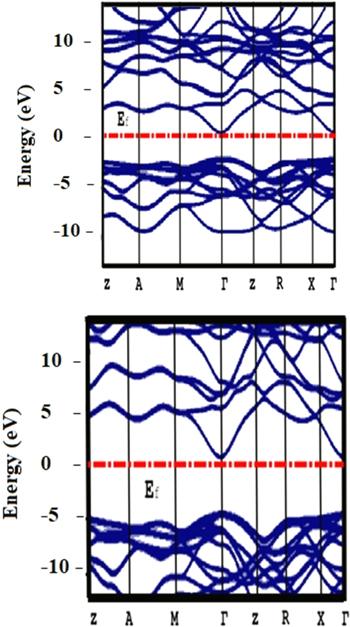
Figure 2. Band structures of a pure rutile GeO2 calculated using the LDA approximation (top) and using the LDA-SIC approximation (bottom). |
Table 2. Comparison between some values for the direct bandgap energy of rutile GeO2 found in the research literature and our values. |
When a semiconductor material interacts with electromagnetic radiation, specific photons may be absorbed by the semiconductor. On the basis of the formula used in [56], Eg (eV)×λ (μm) = 1.24, which relates the wavelength, λ, of the photon and the gap energy of the semiconductor, we calculate the wavelength of the studied rutile GeO2 compound. For the value of 4.80 eV obtained using the LDA-SIC, the corresponding wavelength of the photon energy is 258 nm and the vacuum frequency that matches this wavelength value is 1160.49 THz. This wavelength is within the range of ultraviolet (UV) radiation, which is limited between 200 nm and 380 nm and is visible to birds, insects, and fish. This type of radiation is used in several fields, such as tracking tags on products, bar codes, optical detectors, forensic science, drug screening, protein analysis, DNA sequencing, pharmaceutical research, the medical imaging of cells, and solid-state lighting. Using the LDA approximation, the value of λ obtained is 590 nm, corresponding to a frequency of about 508 THz.
3.2. Half-metallicity and ferromagnetic properties
Half-metals are peculiar ferromagnets that behave like conductors of electrons at the Fermi level in one spin orientation, while their behavior changes to semiconducting or insulating to electrons in the other spin orientation. To examine the presence of this feature in the rutile GeO2 semiconductor, DOS plots were performed. These were obtained by calculating the number of electronic states per unit of energy and per unit cell. The DOS results provide a description of the electronic state distribution in a physical system as a function of electronic energy, which will be considered in this paper using the electron volt unit scale (eV). They can reveal any spin polarization around the Fermi level and the nature of chemical bonds between atoms. In figure 3, we present the total and partial DOS dependence on the energy relative to the Fermi level of pure rutile GeO2 obtained using the LDA-SIC approximation. The black line represents the total DOS, while the other colors show the partial DOSs of each element of the compound. Based on this figure, we can say that no magnetic behavior can be detected if rutile germanium oxide is not doped with impurities. This statement is confirmed by the total magnetic moment, which is found to be zero. From this plot, we can determine the bandgap value, which is equal to 4.80 eV. The latter is consistent with the previous value obtained from the band structure plots.
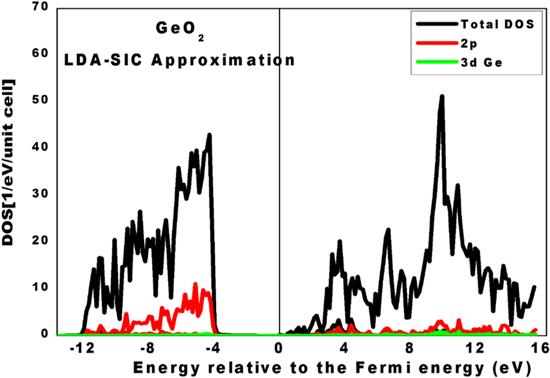
Figure 3. Partial and total DOSs of the pure rutile GeO2 using the LDA-SIC approximation. |
In order to investigate the possibility of enhancing a rutile GeO2 semiconductor with the half-metallicity property, this material was doped with 3d transition metals (Co [Ar] 3d7 4s2, and Fe [Ar] 3d64s2). We set the doping concentration to 10% of the transition metal in all calculations, for the reason explained earlier. The total and partial DOSs of the Ge0.9Co0.1O2 compound were calculated using the LDA and LDA-SIC approximations. The results of doping with Co at a 10% concentration are displayed in figures 4(a) and (b). A spin polarization around the Fermi level is clearly shown in these figures. In the case of figure 4(a), it can be seen that there is no symmetry between the spin-up and spin-down orientations, which means that the Co-doped rutile GeO2 is a magnetic material due to the cobalt atoms injected into the semiconductor. In addition, the specific spin polarization exhibited for the spin-down orientation around the Fermi level is a relevant sign of the presence of the half-metallicity feature in the sample. In figure 4(b), the half-metallicity feature is shown again, in spite of two major differences compared with the result given by the LDA approximation. One can observe a decrease in the strength of the spin polarization at the Fermi level in addition to a change in the sign of the spin polarization. In other words, the correlation effect of electrons reduces the spin polarization. Overall, we can infer, based on these plots, that both theoretical approximations predict a half-metallicity property and magnetic behavior around the Fermi level in the Ge0.9Co0.1O2 compound. We note, however, that the polarization is more significant around the Fermi level in the DOS plots calculated using the LDA approximation.
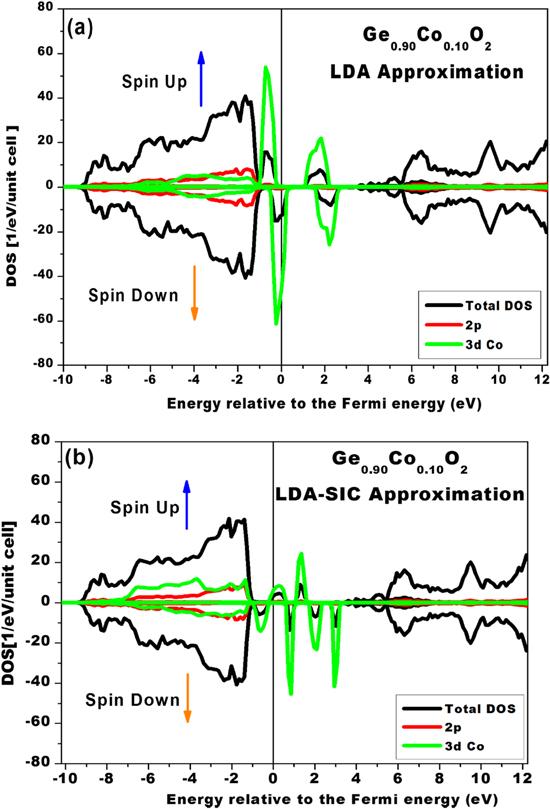
Figure 4. The partial and total DOSs of rutile GeO2 doped with 10% of Co calculated using the LDA approximation (a) and the LDA-SIC approximation (b). |
In conclusion, we can assume, based on the DOS calculations, that 10% Co doping in a rutile germanium oxide semiconductor leads to half-metallic behavior. A possible interpretation of this property is that the 3d layer of Co transition-metal cations mainly occupies cation sites. This leads to the result that the cations are tetrahedrally arranged with p-anions of oxygen. Consequently, p-d hybridization takes place between germanium and the surrounding oxygen atoms, which can be seen in the valence band. Finally, the Ge0.9Co0.1O2 compound can be considered to be a DMS material.
Similar behaviors (i.e. the magnetic behavior and the half-metallicity feature around the Fermi level) in rutile GeO2 doped with 10% of Fe (Z = 26) can be observed in figures 5(a) and (b). Spin polarization around the Fermi level is clearly exhibited in these figures obtained respectively using the LDA and LDA-SIC approximations. Again, the spin polarization is greater for the DOS calculations that used the LDA approximations. Compared to the plots of rutile GeO2 doped with Co, the value of spin polarization obtained using the LDA approximation is slightly greater in the case of Fe doping. However, the spin polarizations seen in the DOS plots calculated using the LDA-SIC approximation are fairly equal for both transition-metal dopants.
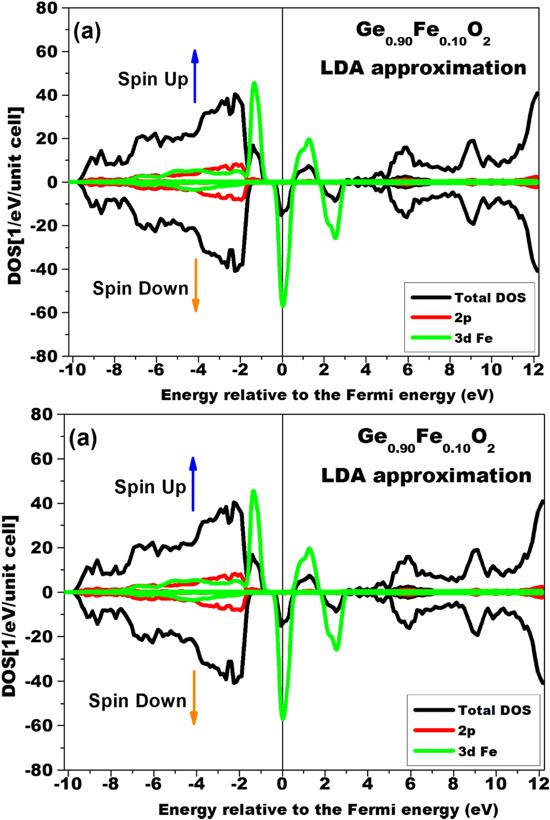
Figure 5. The partial and total DOSs of rutile GeO2 doped with 10% of Fe calculated using the LDA approximation (a) and calculated using the LDA-SIC approximation (b). |
Figures 6(a) and (b) display the behaviors of rutile GeO2 doped with 10% of Ni calculated using the LDA and LDA-SIC approximations. In the first figure 6(a), the compound remains nonmagnetic, as confirmed by two facts: the symmetry between the up and down spins as well as the total moment of the system remain equal to zero; however, it is found that it changes its behavior by becoming a P-type semiconductor instead of the N-type of the pure case, which can be explained by the displacement of the valence band towards the Fermi level. As a result of using the correction given by the LDA-SIC approximation (see figure 6(b)), the system shows (unlike figure 6(a)) magnetic behavior, which is characterized by a half-metallic comportment with a polarization of 100% at the Fermi level.
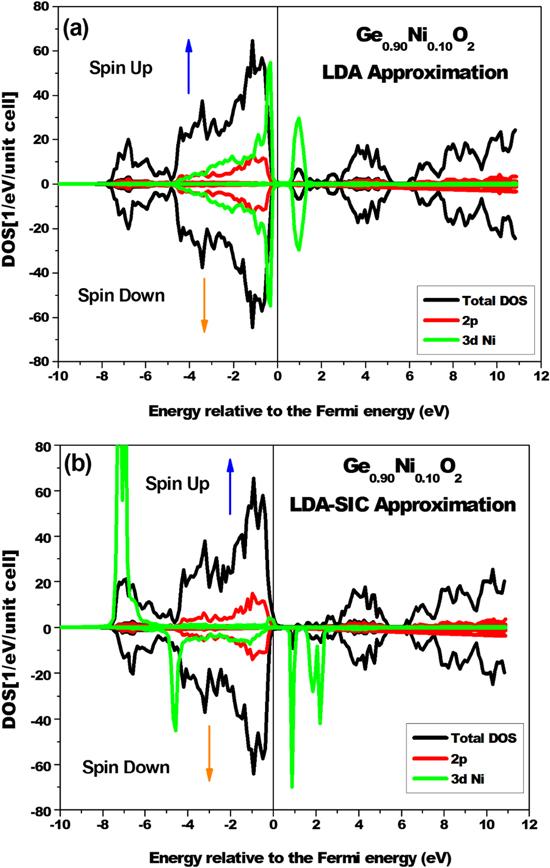
Figure 6. The partial and total DOSs of rutile GeO2 doped with 10% of Ni calculated using the LDA approximation (a) and the LDA-SIC approximation (b). |
The effect of the simultaneous co-doping of GeO2 with Fe (5%) and Co (5%) was examined in this study. Co-doping has the benefit of enhancing the critical temperature and the magnetic momentum around the Fermi level. Calculations of the total and partial DOSs of a Ge0.9Fe0.05Co0.05O2 compound were performed using the LDA and LDA-SIC approximations. The results are given in figures 7(a) and (b). The persistence of half-metallicity can be observed in the co-doped rutile GeO2. In addition, we noticed an enhancement of the spin polarization around the Fermi level in the results obtained using the LDA-SIC approximation. These results can be interpreted as a consequence of a p-d interaction between the p electrons of oxygen atoms and the d electrons of neighboring Co and Fe atoms. This interaction is combined with the d-d interaction due to the presence of the d electrons of Co and the d electrons of Fe. On the other hand, in the cases of Fe and Co, Fe and Ni, and Co and Ni, there is no indication of charge transfer between these atoms. The Fe2+ and Co2+, Ni2+ and Co2+, and Fe2+ and Ni2+ valence configurations are limited in the (Fe, Co)/(Ni, Co)/(Fe, Ni) doped GeO2. As a result, a gain in kinetic energy due to the jump of spin-polarized carriers does not seem to occur between these ions; as a result, the double exchange mechanism is not effective in the co-doped compound. This case is similar to that previously reported in [57] for the Zn0.88Fe0.06Co0.06O system.
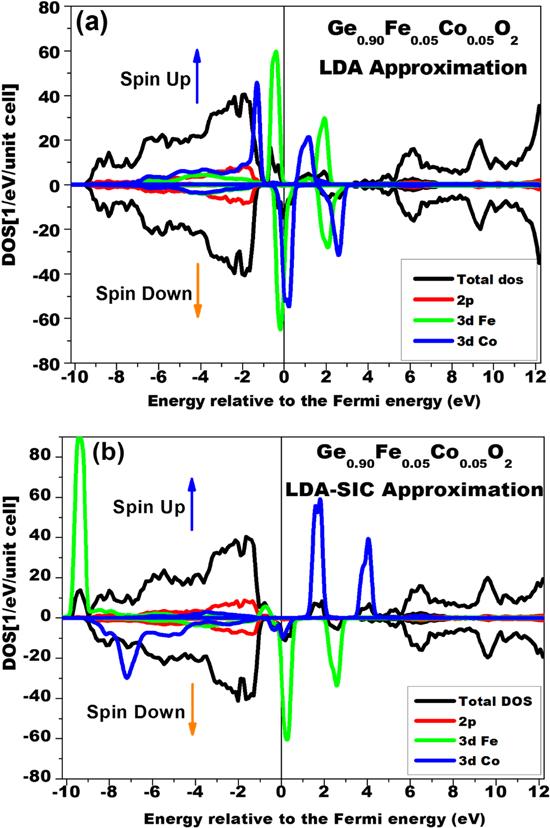
Figure 7. Partial and total DOSs of rutile GeO2 co-doped with 5% of Fe and 5% of Co calculated using the LDA approximation (a) and the LDA-SIC approximation (b). |
The effect of co-doping GeO2 with Ni (5%) and Co (5%) is observed in figures 8(a) and (b), which were produced using the LDA and LDA-SIC approximations. In the first case, in figure 8(a), the behavior is half-metallic due to the location of spin-downs at the Fermi level, which is always the consequence of p-d interaction, while in figure 8(b), which shows the results of the calculation using the LDA-SIC approximation, although the compound is magnetic, it presents a metallic behavior, since there are peaks at the Fermi level for the up and down spins.

Figure 8. Partial and total DOSs of rutile GeO2 co-doped with 5% of Ni and 5% of Co calculated using the LDA approximation (a) and the LDA-SIC approximation (b). |
Figures 9(a) and (b) display the simultaneous effect of co-doping GeO2 with Ni (5%) and Fe (5%) using both the LDA and LDA-SIC approximations as corrections. When Fe and Ni are used to co-dope GeO2, according to the LDA approximation as shown in figure 9(a), the system is magnetic and presents a half-metallic behavior, while according to the LDA-SIC approximation as shown in figure 9(b), the matrix is not magnetic, which may be inferred from the existence of symmetry between spin-up and spin-down.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9. Partial and total DOSs of rutile GeO2 co-doped with 5% of Ni and 5% of Fe calculated using the LDA approximation (a) and the LDA-SIC approximation (b). |
3.3. Critical temperature
To calculate the critical temperature TC values of the studied DMS material, the energy difference ΔE between EDLM and EFM (which respectively denote the energy of the disordered local moment state (DLM) and the energy ferromagnetic state) should be determined. Once EDLM and EFM are evaluated using the frameworks of the LDA and LDA-SIC approximations, the Curie temperature TC can be obtained using the formula [58]:
$\begin{eqnarray*}{T}_{C}=\displaystyle \frac{2}{3k}\cdot \displaystyle \frac{{\rm{\Delta }}E}{C},\end{eqnarray*}$
where ΔE = EDLM − EFM is the energy difference, $C$ is the doping concentration and $k$ is the Boltzmann constant.In table 3, we display the values of the critical temperature TC for the different cases treated. Based on these results, we conclude that Fe doping of the rutile GeO2 semiconductor leads to Curie temperature values that are more consequential than those of the Co-doped compound. This conclusion is verified for both the LDA and LDA-SIC approximations. It should be mentioned that the calculated values for the Fe dopant are greater than room temperature, which is important for practical DMS materials. On the other hand, we can see that co-doping enhances the critical temperature and that the value of TC reaches 398K when calculated using the LDA-SIC approximation. We believe that the strong p-d and d-d interactions contribute to the occurrence of ferromagnetism in Ge0.90Co0.05Fe0.05O2. This makes the rutile GeO2 semiconductor a promising candidate for incorporation into spintronic devices.
Table 3. Values of the critical temperature TC calculated using the LDA and LDA-SIC approximations for rutile GeO2 doped with 10% of Co, 10% of Fe, and co-doped with 5% of Co and 5% of Fe. |
| Rutile GeO2 | Critical temperature (in Kelvin) | |
|---|---|---|
| LDA approximation | LDA-SIC approximation | |
| Doped with 10% of Co | 230 | 190 |
| Doped with 10% of Fe | 350 | 334 |
| Co-doped with 5% Co and 5% Fe | 380 | 398 |
| | ||
| Rutile GeO2 | Critical temperature (in Kelvin) | |
| | ||
| LDA approximation | LDA-SIC Approximation | |
| | ||
| Doped with 10% of Ni | — | 88 |
| Co-doped with 5% Ni and 5% Fe | 210 | — |
| Co-doped with 5% Ni and 5% Co | 125 | 75 |
Table 4. Values of the formation energies calculated using the LDA and LDA-SIC approximations for rutile GeO2 doped with 10% of Co, 10% of Fe, and co-doped with 5% of Co and 5% of Fe. |
| Formation energies (in Rydberg) | ||
|---|---|---|
| Rutile GeO2 | LDA approximation | LDA-SIC approximation |
| Doped with 10% of Co | −0.707 | −0.807 |
| Doped with 10% of Fe | −0.787 | −0.877 |
| Co-doped with 5% Co and 5% Fe | −0.657 | −0.922 |
| Doped with 10% of Ni | −0.522 | −0.612 |
| Co-doped with 5% Ni and 5% Fe | −0.536 | −0.677 |
| Co-doped with 5% Ni and 5% Co | −0.423 | −0.789 |
4. Formation energies
In order to investigate the stabilities of all the studied compounds, the formation energy ΔEf was calculated using the two approximations, LDA and LDA-SIC (See Table 4). This energy is defined in the first instance as the difference in total energy between the doped compounds Ge1-xFexO2 or Ge1-xCoxO2 and GeO2 and in the second instance as the difference in total energy between the co-doped compounds Ge1-2xFexCoxO2 and the doped Ge1-xFexO2 or Ge1-xCoxO2. The calculated results showed negative formation energies, indicating the stability and thermodynamic feasibility of these compounds.
5. Conclusions
In this work, the possibility of using doped or co-doped rutile GeO2 as a DMS material was explored. Doping or co-doping by replacing this material with Co Ni and Fe transition metals was investigated because of the d-layer magnetic impact of such atoms. The doping concentration was set to 10% to avoid the percolation threshold (16.66%). To fulfil the objectives of this research, the half-metallicities and critical temperatures of doped and co-doped rutile GeO2 compounds were theoretically investigated. The semiconductor natures and bandgap values of these compounds were determined using calculations of their band structures. The band structure results show a direct ultrawide bandgap at the Γ point, which makes these compounds good candidates for optoelectronic applications. On the other hand, the DOS curves clearly confirm the half-metallicity of rutile GeO2 doped or co-doped with 3d transition metals such as Fe, Ni, and Co, even though this property is more visible in the DOSs calculated using the LDA approximation. Furthermore, the co-doping of this compound slightly enhances the spin polarization around the Fermi level and visibly improves the critical temperature. Finally, one can conclude that rutile GeO2, which is thermodynamically stable, is a good candidate for utilization as a half-metallic compound in spintronic devices. The rutile germanium oxide DMS material also has the potential to be incorporated into some optoelectronic devices.