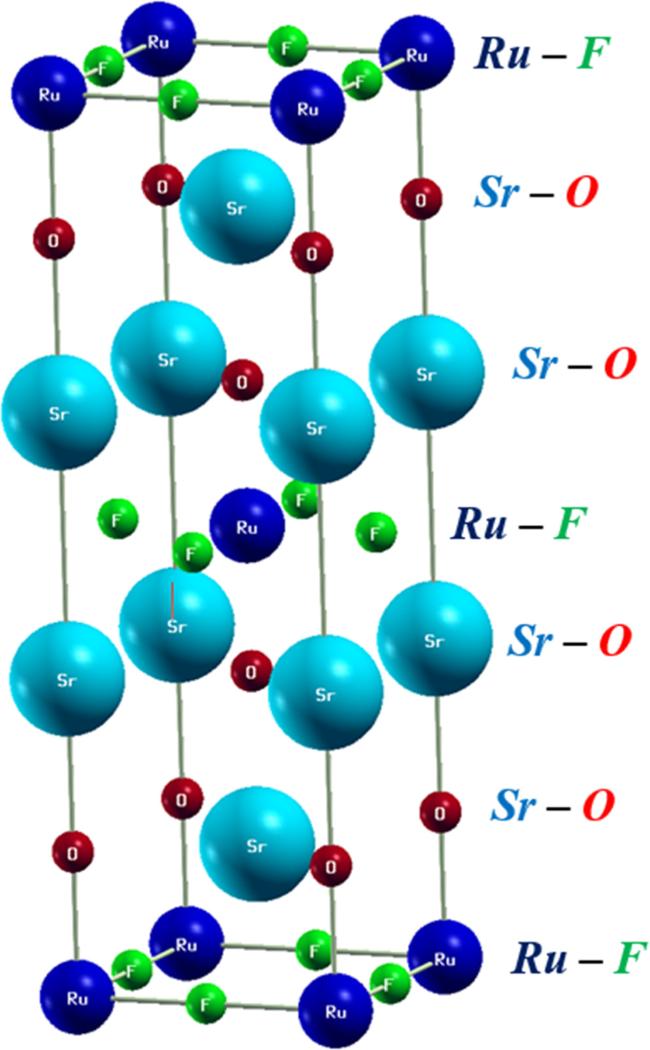
Regarding the quaternary alloy Sr
2RuO
2F
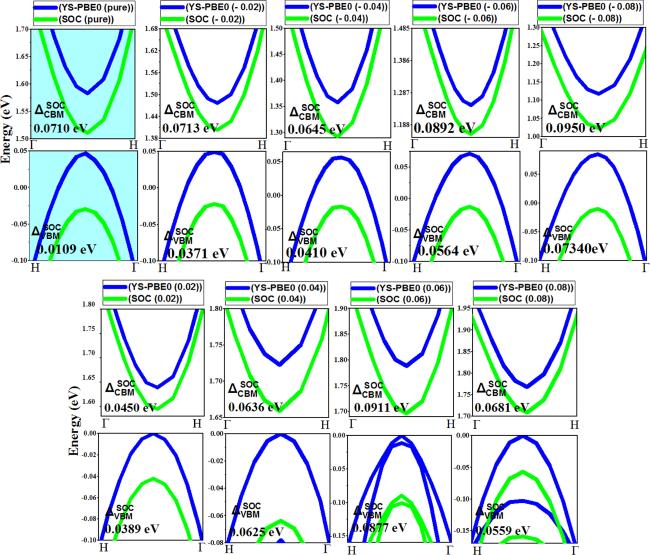
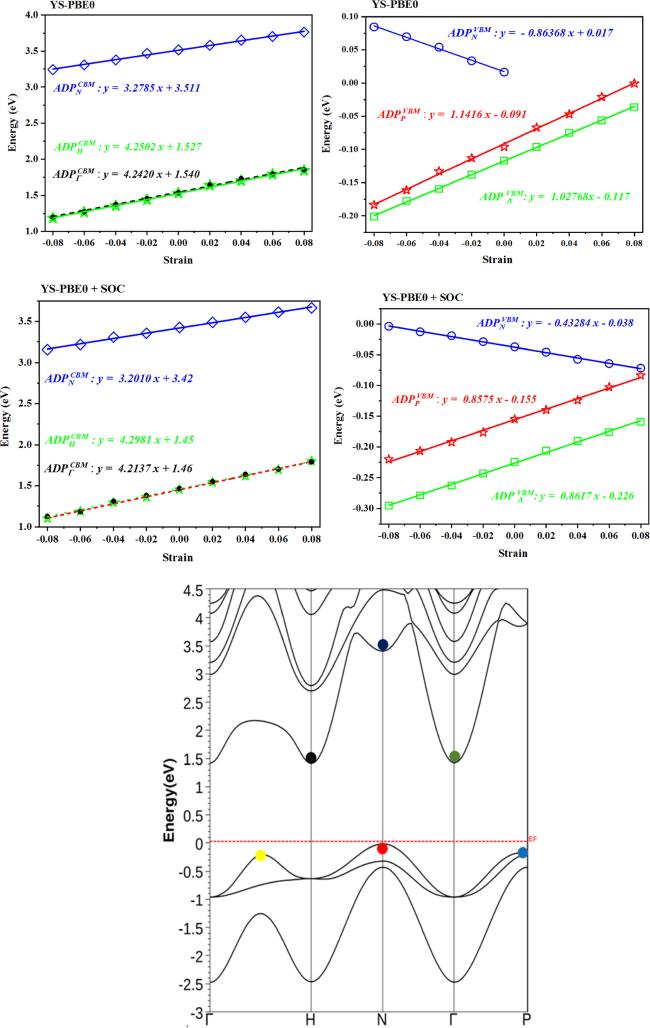
2, the state at the N point of the valence band (VBM) exhibits metal-like characteristics and has a bonding interaction with the F and O orbitals. As the alloy is expanded, the energy of this state experiences a significant decrease using both the YS-PBE0 approach and SO coupling. This is due to the fact that as the distance between atoms increases, the bond length (i.e.
stretching) also increases, leading to a decrease in energy. The bonding nature of the state contributes to an increase in energy when the bond length shortens, but as the bond length increases, the energy also decreases. (
see figure
6). The kinetic energy of electrons within a substance is intimately related to the motion of these subatomic particles. As the distance between atoms within a compound expands (i.e.
bond length under stretch), the energy of these electrons correspondingly decreases due to the greater spatial separation between atoms. This reduction in energy is a direct consequence of the decline in the momentum of the electrons as the atoms are farther apart, and the opportunities decline in the momentum of the electrons as the atoms are farther apart, and the opportunities for interaction with the nuclei are accordingly diminished. The correlation between the potential energy of electrons in a material and the electrostatic interactions between these electrons and nuclei is a complex and fascinating phenomenon. As the distance between nuclei, otherwise known as bond length, increases, the potential energy of the electrons correspondingly rises as a result of the weakening of the electrostatic interactions. This highlights the intricate interplay between the various components of Sr
2RuO
2F
2 quaternary alloy and their effect on the overall energy state. The kinetic energy of the electrons decreases at a greater rate than the increase in potential energy, resulting in a net diminution of the aggregate energy of the system. This salient phenomenon, intrinsic to the bonding state at the valence band maximum (VBM), is the very explanation for the diminutive ADP observed in table
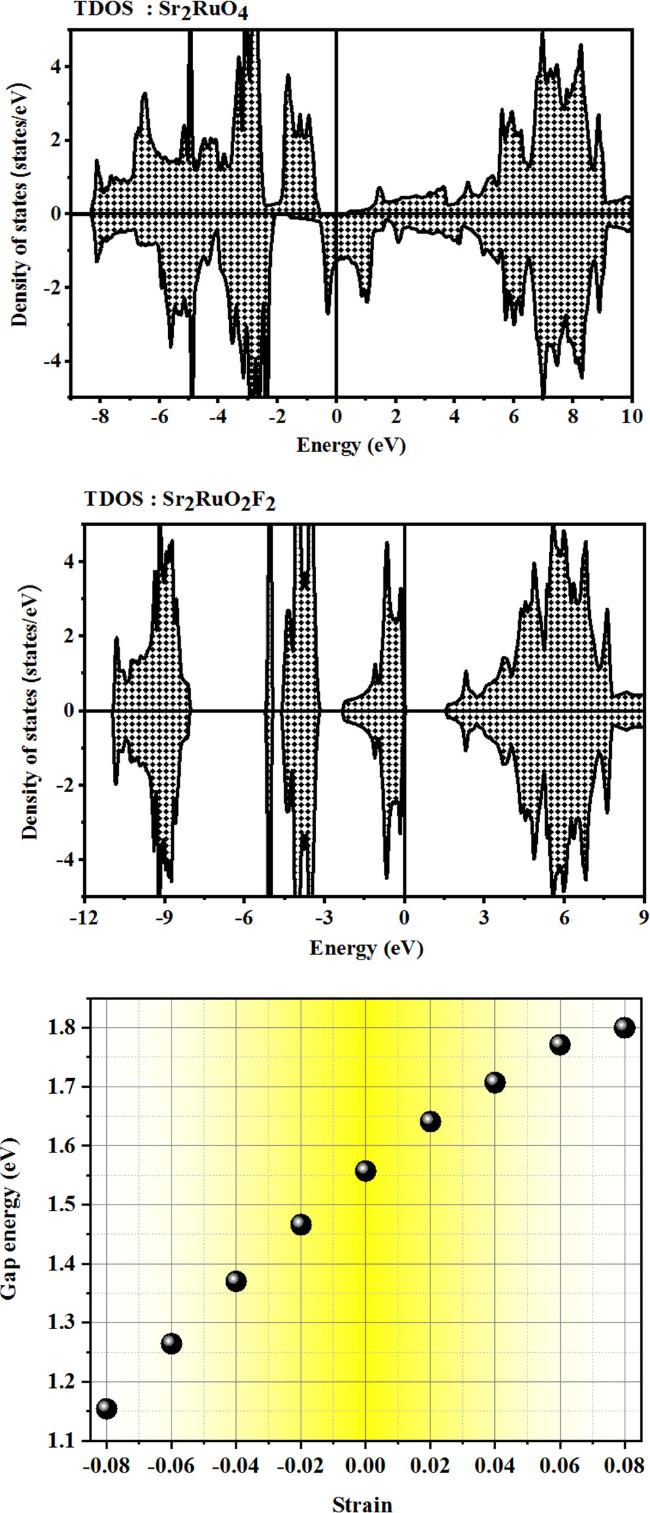
2. In general terms, the effect of antibonding interactions on the energy edges of a material can be significant, particularly in the conduction band (CBM). As mentioned earlier, the antibonding states have a lower electron density between the nuclei, which leads to higher repulsion and increased energy. This elevation of energy can propel the state into the conduction band, thereby rendering it amenable for electrical conduction [
36]. In Sr
2RuO
2F
2 semiconductor, the antibonding interactions arise from the overlap of the electronic wave functions of the Ru-
4d orbitals in the RuO
2 planes with the F-2
p orbitals in bonds and the electrons of the F layers. The antibonding interactions are formed between the electrons of the Ru–O and the Ru–F bonds. The Born–Oppenheimer approximation is a fundamental principle in quantum mechanics that explains the relationship between the positions of nuclei and electrons in a substance, and the resulting energy levels [
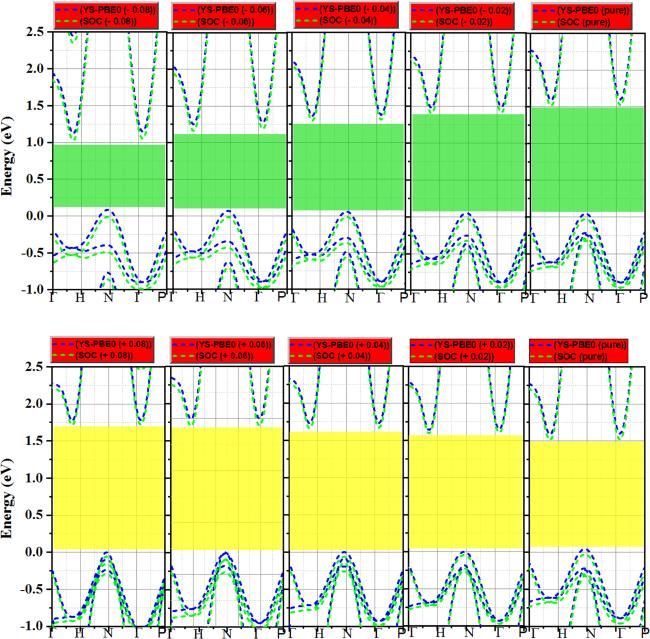
37]. According to this principle, when the bond length lengthens (e.g.
from ϵz = −0.08
to ϵz = + 0.08), the energy of the antibonding states at
N,
H and Γ points also rises, as shown in figure
6. This is because the electrons in these antibonding states, which have higher energy than the bonding states, are farther apart, resulting in higher potential energy. As illustrated in figure
6, the conduction band (CBM) electrons are more sensitive to external strains/deformations, and their energy levels change more readily in response to these changes. The energy edge is significantly affected by strain; it increased steadily under pressure/stretch and abruptly jumped to ≈3.77 eV, ≈1.87 eV and ≈1.85 eV at N, H, and Γ points respectively, when
ϵz = + 0.08 (
see figure
6). This results in the ADP being larger in the conduction band than in the valence band, as listed in table
2. The previously observed behavior persists even upon applying SO coupling.




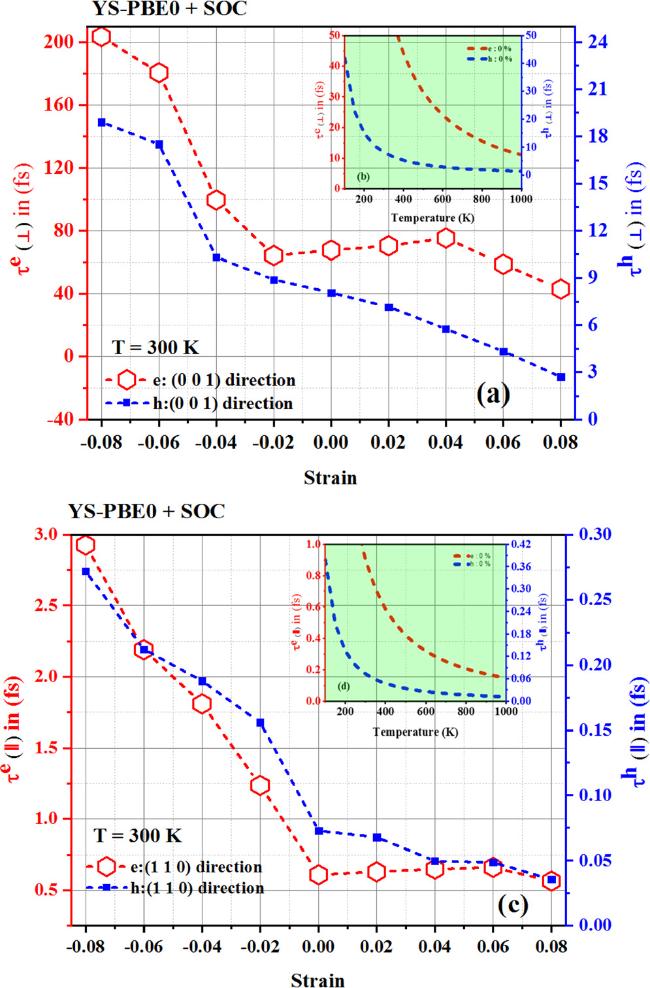
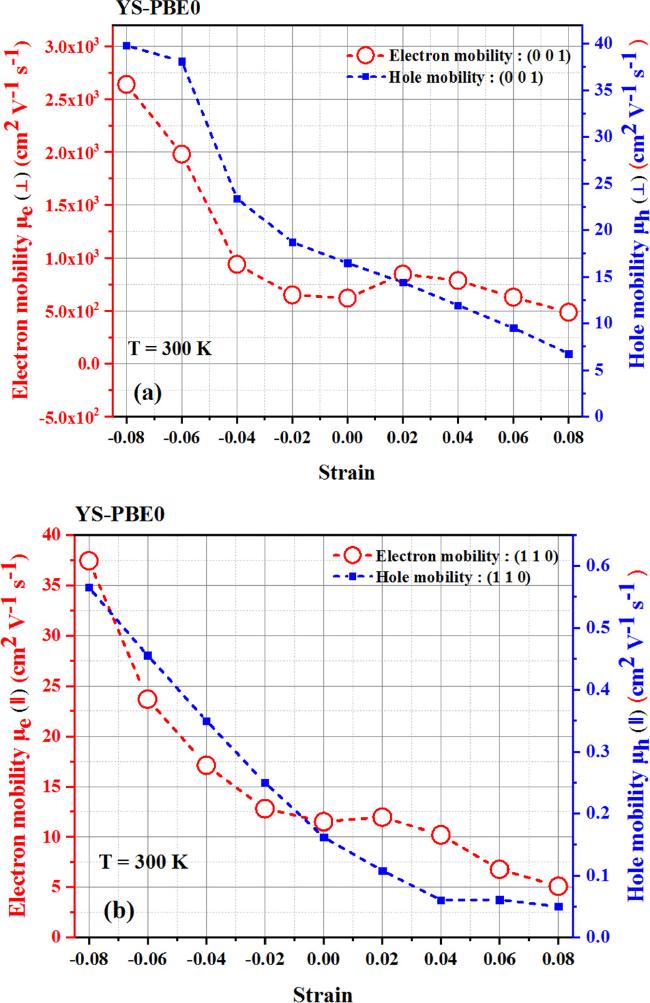
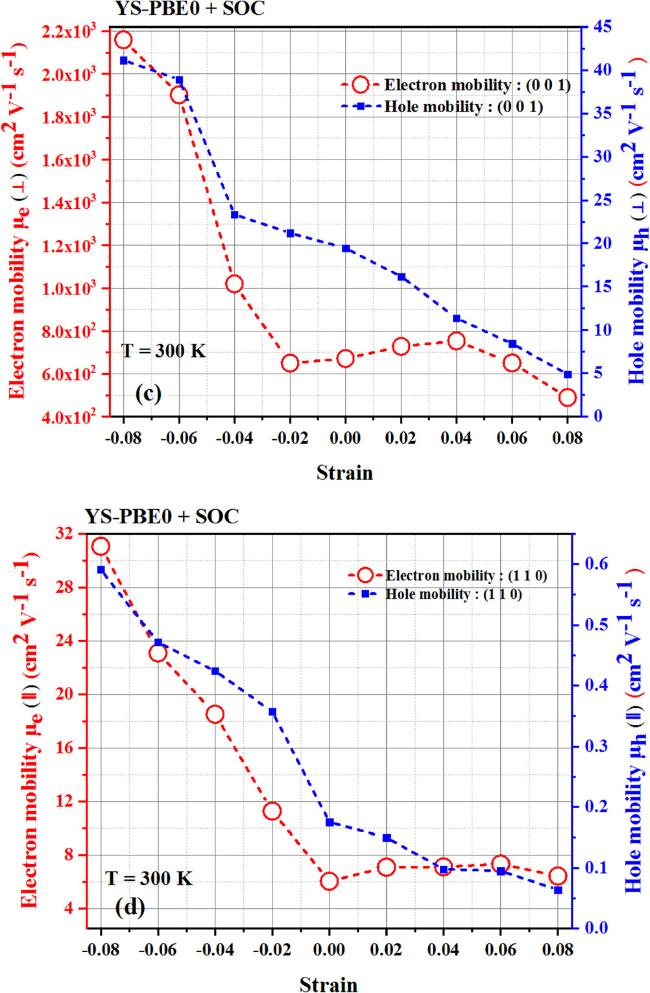

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}