



Introduction
Nickel-based single-crystal superalloys are widely used in manufacturing turbine blades in aerospace engines due to their excellent high-temperature resistance to fatigue, creep, and oxidation [1–5]. Numerous solute atoms (e.g., Al, Ta, W, Mo, Cr, Co, Re, and Ru) were introduced into the nickel-based single-crystal superalloys [6–8]. They play roles in stabilizing the alloy structure, increasing the volume fraction of γ′ phase, strengthening γ and γ′ phases, inhibiting the precipitation of harmful phases, and resisting oxidation [9–13]. Adding Mo will cause the local lattice distortion of the γ phase, generating a large stress field and hindering the movement of dislocations, thereby achieving a solid solution-strengthening effect [14–17]. The Ru element can suppress the precipitation of harmful phases in the matrix, reducing the occurrence of cracks and thereby improving the high-temperature structural stability of the alloy [18–21].
Solute atoms usually exist in the nickel lattice as isolated single atoms. There may be an intermediate state, solute clusters, between solute atoms [22]. So far, fewer reports have systematically studied the behavior characteristics and microscopic mechanism of solute clustering in nickel-based single-crystal superalloys. The strengthening from the cluster may be an exaggerated form of solid solution strengthening, instigated by interactions between solutes and dislocations [23, 24]. Firstly, clusters can serve as impediments to the motion of dislocations within the crystal lattice, impeding their mobility and consequently reinforcing the material's strength. Additionally, specific clusters, such as coherent precipitates, can obstruct the movement of dislocations and grain boundaries, thereby bolstering the creep resistance of the superalloy. It is significant to study the possibility and distribution characteristics of atomic clusters in nickel-based superalloys and understand the relationship between solute clustering and alloy stabilizing, thus providing a theoretical basis and process guidance for creating high-temperature alloys with extended lifespans and heightened reliability.
First-principles calculations have emerged as a prevalent tool for investigating the characteristics of phases and interfaces within nickel-based single-crystal superalloys. Through density-functional theory (DFT) simulations, thermodynamic parameters encompassing site preference, segregation tendencies, and separation energies of alloying elements can be derived, thereby facilitating an interpretation of the influence of alloying on thermodynamic stability [25–27]. The reliability of the DFT methodology has been corroborated through experimental investigations employing three-dimensional atom probe tomography, validating its accuracy and efficacy in modeling alloy behavior [28–30].
Herein, the first-principles calculations were employed to investigate the possibility of the solute cluster and explore the stabilizing effect of the cluster on the γ phase of the nickel-based single-crystal superalloy. The γ phases with Mo−Mo and Mo−Mo−Ru clusters are built as models to calculate. Note that the γ phase predominates as the primary detrimental phase for dislocation propagation during creep. Augmented stability of the γ phase may reinforce the creep resistance of high-temperature alloys. Adding Mo can reduce the total energy, binding energy, and formation energy of the γ phase by replacing weak Ni−Ni interaction with strong Mo−Ni bonding. It is worth noting that the γ phase with the Mo−Mo cluster is more stable than that with the Mo single atom, possibly due to a wide affecting range. The Ru atom further stabilizes the γ phase and preferentially forms the Mo−Mo−Ru cluster rather than existing in the γ phase in isolation. The stabilizing mechanism of the Mo−Mo−Ru cluster may be the substitution of weak Ni−Mo interaction with strong Ru−Mo interaction, probably originating from the enhanced d-band hybridization.
Methods
Density-functional theory calculations were performed using the Vienna Ab initio Simulation Package (VASP). The exchange–correlation energy was described using the Perdew–Burke–Ernzerhof (PBE) parametrization of the generalized gradient approximation (GGA) method. The cut-off energy of 400 eV was chosen for the plane wave basis. A 3 × 3 × 3 Monkhorst–Pack k-point mesh was used for Brillouin zone integrations. The energy and forces were converged to 10−5 eV and −0.02 eV Å. The models of γ phases were built using 108 atom (3 × 3 × 3 supercells). The binding energy was calculated using the equation: Eb = E − $\displaystyle \sum _{i}{E}_{{\rm{atom}}}(i),$ where E is the total energy, Eatom (i) refers to the energy of the free atom. The formation energy was determined using the formula: ΔE = (E − $\displaystyle \sum _{i}{n}_{i}{{\mu }}_{i}$) /$\displaystyle \sum _{i}{n}_{i},$ where E represents the total energy, ${\mu }$i is the chemical potential of i element, ni is the number of i element. Following the attainment of full self-consistency in electronic-structure calculations, an energy-partitioning technique, Crystal Orbital Hamilton Population (COHP), is employed to dissect the overall band-structure energy into distinct interatomic interactions. The lobster script was used to analyze COHP.
Results and discussion
Mo segregates in the γ phase and plays a solid solution-strengthening role in nickel-based superalloys. To clarify the possibility of a Mo−Mo cluster in the γ phase, we built γ phase models, including Ni108, Ni107Mo, and Ni106Mo2, as shown in figures 1(a)–(c). Total energy denotes the energy state of the system when atoms are positioned ideally within the crystal lattice. Binding energy signifies the energy required to separate the atoms within the crystal to an infinite distance. Formation energy refers to the energy required to create a specific defect, compound, or structure from its constituent elements in their most stable states. Low total energy, binding energy, and formation energy of the system indicate a stable structure. It is calculated that the total energy, binding energy, and formation energy (figures 1(d)–(f) conform to the order of Ni106Mo2 < Ni107Mo < Ni108, indicating the introduction of Mo can improve the system stability. Note that the energies of Ni106Mo2 are the lowest, which confirms the Mo−Mo cluster is more likely to exist in the γ phase, and they can stabilize the matrix.
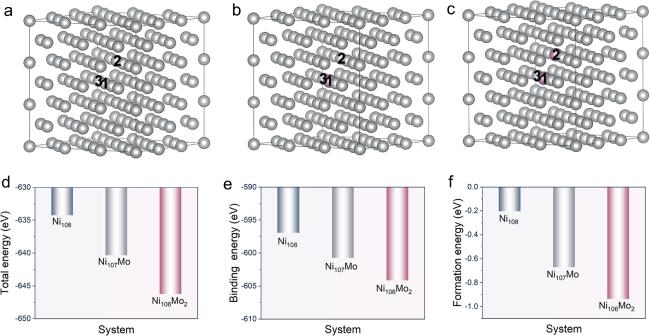
Figure 1. The γ phase models of (a) Ni108, (b) Ni107Mo, and (c) Ni106Mo2 after structural optimization. Gray and purple balls represent Ni and Mo atoms, respectively. (d) Total energies, (e) binding energies, and (f) formation energies for diverse models. |
The system stability enhancement may be attributed to the intensified interaction between atoms. The interaction between Mo (or Ni in the same position) and the nearest neighboring (NN) Ni atom was investigated using the projected and integrated crystal orbital Hamilton population (pCOHP and iCOHP). As displayed in figure 2, the iCOHP follow the sequence of Ni107Mo < Ni106Mo2 < Ni108 between atoms 1 and 3, Ni106Mo2 < Ni108 < Ni107Mo between atoms 2 and 3. It suggests that the Ni−Mo interaction is stronger than the Ni−Ni interaction, possibly due to the more unpaired electrons of Mo than Ni. Moreover, the Mo single atom and Mo−Mo cluster can intensify the interaction with the NN Ni atom, and the cluster possesses a wider affecting range than the single atom. Accordingly, the enhanced effect of clusters on system stability may arise from the strong Ni−Mo interaction and more Ni interacting with Mo. It is noted that the intensified interaction among atoms may facilitate a decrease in the diffusion rate of matrix elements, thereby contributing to solid solution strengthening.
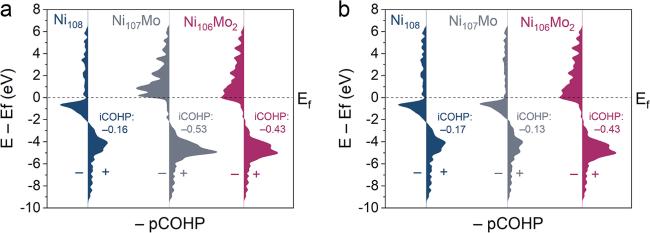
Figure 2. (a) The pCOHPs between atom 1 and 3 in different γ phases. (b) The pCOHPs between atom 2 and 3 in different γ phases. |
Ru can alter the distribution of alloy elements, suppressing the formation of topologically close-packed (TCP) phases. Note that Ru atoms possess more unpaired electrons than Ni atoms, potentially resulting in a stronger Ru−Mo interaction relative to Ni−Mo, thereby improving the system stability. To study the relationship between the Ru atom and the Mo−Mo cluster, we built the γ phases including a Ru atom at the nearest and second neighboring (SN) positions of the Mo−Mo cluster (figures 3(a)–(c)). The total energy, binding energy, and formation energy (figures 3(d)–(f) follow the order of Ni105Mo2Ru (NN) < Ni105Mo2Ru (SN) < Ni106Mo2, which suggests that the introduction of the Ru atom can heighten the system stability further. Additionally, the Ru atom preferentially occupies the NN position rather than the SN position of the Mo−Mo cluster. This means the Ru atom is inclined to form a Mo−Mo−Ru cluster other than existing in the γ phase in isolation.
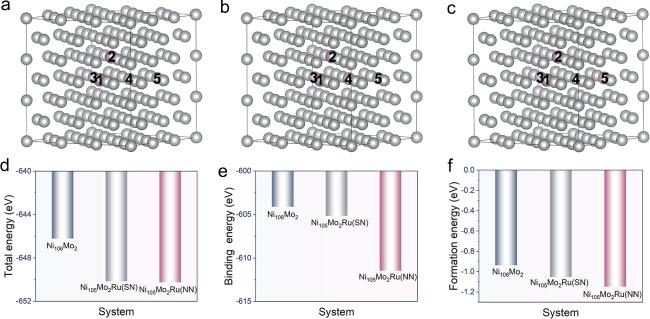
Figure 3. The γ phase models of (a) Ni106Mo2, (b) Ni105Mo2Ru (NN), and (c) Ni105Mo2Ru (SN) after structural optimization. Gray, purple, and brown balls represent Ni, Mo, and Ru atoms, respectively. (d) Total energies, (e) binding energies, and (f) formation energies for diverse models. |
The bond length, COHP, and electron localization function (ELF) analyses were carried out to explore how the Ru atom influences Mo−Mo bonding. The Mo−Mo bond length (figure 4(a)) exhibits the order of Ni106Mo2 < Ni105Mo2Ru (SN) < Ni105Mo2Ru (NN), possibly corresponding to a strength sequence of Ni106Mo2 > Ni105Mo2Ru (SN) > Ni105Mo2Ru (NN). As presented in figure 4(b), the iCOHP between two Mo atoms shows the order of Ni106Mo2 < Ni105Mo2Ru (SN) < Ni105Mo2Ru (NN), in line with the inference of the Mo−Mo bond length. Furthermore, the maximum ELF values (figures 4(c)–(e)) occur near the midpoint of the Mo−Mo bond, and they display an order of Ni106Mo2 > Ni105Mo2Ru (SN) > Ni105Mo2Ru (NN), coinciding with the deduction above. These results reveal that introducing Ru may weaken the Mo−Mo interaction. Moreover, the close distance between the Ru atom and Mo−Mo cluster leads to weak Mo−Mo bonding.
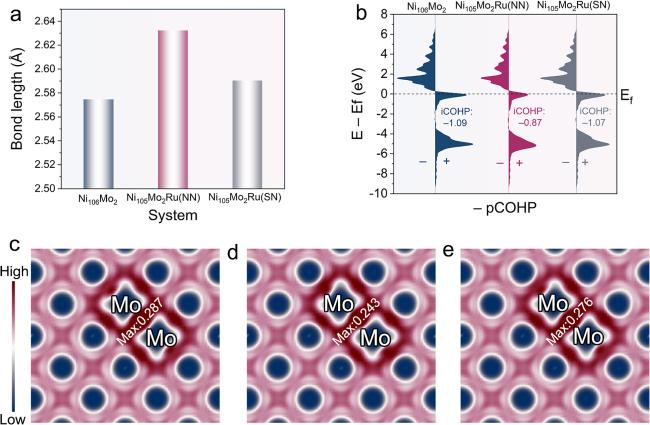
Figure 4. (a) The Mo−Mo bond lengths. (b) The pCOHPs between atom 1 and 2. The ELF plots along (010) plane for (c) Ni106Mo2, (d) Ni105Mo2Ru (NN), (e) Ni105Mo2Ru (SN). |
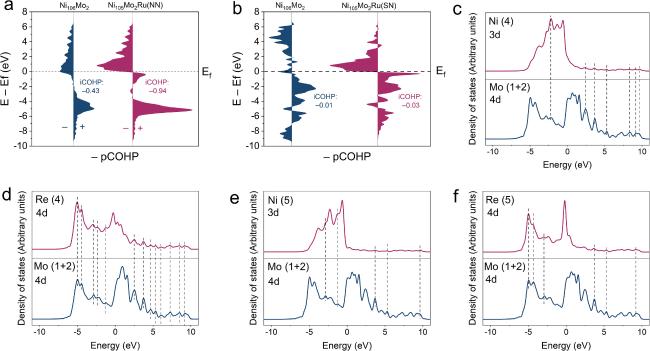
The Ru−Mo interaction was explored using COHP and partial density of states (PDOS). As can be seen in figures 5(a) and (b), the iCOHP value between atoms 2 and 4 in the Ni105Mo2Ru (NN) is lower than that in the Ni106Mo2. Additionally, the atoms 2 and 5 in the Ni105Mo2Ru (SN) deliver a more negative iCOHP value than that in the Ni106Mo2. These results indicate that the Ru−Mo interaction is stronger than the corresponding Ni−Mo interaction. The PDOS can explain the above phenomenon. As depicted in figures 5(c)–(f), whether the Ru atom is at the NN or SN position of Mo−Mo clusters, the resonance peaks between Ru 4d and Mo 4d are more than those between Ni 3d and Mo 4d. It suggests the stronger Ru−Mo interaction than the Ni−Mo interaction may be due to increased d-orbital hybridization degree.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. (a) The pCOHPs between atoms 2 and 4. (b) The pCOHPs between atoms 2 and 5. (c−f) PDOS of the atoms in Ni106Mo2, Ni105Mo2Ru (NN), and Ni105Mo2Ru (SN). |
Conclusion
In this work, the first-principles calculations were performed to research the possibility and stabilizing mechanism of solute clusters in the γ phase. Introducing Mo can decrease the energies of the γ phase by substituting weak Ni−Ni interaction with strong Mo−Ni bonding. Possibly due to a wide affecting range, the γ phase with the Mo−Mo cluster is more stable than that with a Mo single atom. The Ru solute can further reduce γ phase energies and preferentially form Mo−Mo−Ru clusters. Note that the reason for system stabilization is the replacement of weak Ni−Mo interaction by strong Ru−Mo interaction, likely stemming from the intensified d-orbital hybridization. This observation offers insights into the underlying mechanisms driving solute aggregation behavior and establishes a relationship between clustering behavior and system stability.