



1. Introduction
The ion-matter interaction has been a fundamental and significant research topic in the field of nuclear science and technology. Its diverse applications in nuclear engineering [1, 2], nuclear detection [3], material modification [4, 5], and ion therapy [6, 7] have attracted considerable attention. The underlying mechanisms of ion-matter interactions are being studied, including the investigation of the physical and chemical effects induced in materials by ion irradiation. As ions traverse in materials, ion energy is lost due to the decelerating force exerted by the host atoms. This energy loss is quantified by the stopping power (S), representing the energy transferred from the projectile to the target per unit path length traversed [8, 9]. The S can be grouped into two categories: electronic stopping power (Se), which is caused by inelastic collisions between ions and electrons that lead to electronic excitation and ionization, and nuclear stopping power Sn, which is attributed to atomic displacement from elastic collisions between ions and host nuclei [10, 11]. The Se of materials has been studied since the beginning of the last century [12, 13]. However, the study of the electronic energy loss remains challenging because of its quantum mechanical properties [14–16].
Many theories and models have been proposed to calculate the Se of ions and to investigate the underlying mechanisms responsible for the electronic energy loss [17–19]. The Coulomb scattering formulas [20–22] provide an important theoretical foundation and experimental basis for the study of interactions between particles and materials. Additionally, these formulas demonstrate that the collision parameter plays an important role in ion-matter interactions. The Bethe-Bloch formula describes the Se due to inelastic collisions of fast charged particles with solids [23],
$\begin{eqnarray}{S}_{{\rm{e}}}=\frac{4\pi {z}^{2}{e}^{4}NZ}{{m}_{0}{v}^{2}}{\rm{ln}}\left(\frac{2{m}_{0}{v}^{2}}{I}\right),\end{eqnarray}$
where N is the atomic density of the target materials, and I denotes the mean excitation energy of target electrons. It is concluded that the Se is proportional to the square of the charges of the projectiles (z) and also to the atomic number of the target materials (Z). The free electron gas (FEG) model demonstrates that the Se of slow ions is proportional to the projectile velocity, which can be expressed as Se = Q(Z1,rs)v [24], where Q represents the friction coefficient of the FEG model, which depends on the atomic number of the projectiles (Z1) and the Wigner-Seitz radius (rs) of the FEG. Within the linear response theory, the FEG model [25] assumes constant effective electon density, and this approach is only applicable to the high-energy case, as it fails to consider the charge capture behavior of the projectiles in the low-energy range (v < 1 au, 1 au = 21.9 Å fs−1). Echenique et al [26] calculated the Se of the FEG model by a nonlinear density-functional theory (DFT) approach. This method is used to characterize the response of matter to strong perturbations by taking into account higher-order perturbation terms Nevertheless, most of these methods are based on the FEG model, which ignores atomic and electron band structure of the host materials, leading to discrepancies between the obtained calculations and experimental data. Recently, the real-time time-dependent density functional theory (RT-TDDFT) was employed to study the electronic energy loss of ions, enabling accurate calculations of nonadiabatic energy transfer between ions and electrons, as well as reliable predictions of the Se of materials for ions [27, 28].Recent measurements displayed deviations from the velocity-proportional Se of transition metals for slow protons and helium ions. The stopping cross section (SCS) of Cu [19, 29], Au [19, 29], and Ag [19] for slow protons deviates from the velocity linearity due to d-electron excitation at 0.175, 0.19, and 0.2 au, respectively; while the SCS of helium ions exhibits similar deviations around 0.2 au in these metals. Halliday et al [30] modeled the Se of hydrogen ions in graphite using TDDFT, and provided new insights into the effect of anisotropy of the graphite structure on the Se. It is found that the anisotropic crystal structure and impact parameter possess a strong effect on the Se at low velocities. For projectiles traveling along the graphite planes, a change in the slope of the Se is observed when the projectile velocity reaches the Fermi velocity of the electrons [30]. The Se of Mg for protons and He ions was investigated by using the TDDFT [31], and the deviation from the velocity proportionality was revealed for slow He ions. This deviation is attributed to the electronic structure of He ions due to the charge exchange that opens an additional energy dissipation pathway.
At high velocities, the effect of semicore electron excitation on the Se of Al film for He+ ions was investigated by combining the time-dependent density functional theory method with molecular dynamics simulations (TDDFT-MD) [32]. It was reported that the contribution of semicore electron excitation to the Se is significant for high-velocity protons. First-principles calculations of the Se of water molecules demonstrated that oxygen K-shell excitation is the critical mechanism governing Se beyond the stopping maximum [33]. TDDFT simulations [34] clarified that f-electron excitation of Pt contributes significantly to the Se in the high-energy region under the off-channeling geometry. Moreover, the off-channeling Se including f-electron excitation is in close alignment with the experimental data. TDDFT simulations of iron under ion irradiation [35] revealed that the electrons of iron with n ≥ 3 play a dominate role in the Se for protons and helium ions across a broad velocity regime. These results indicate that the contribution of core-electron excitation should be considered in theoretical studies to enhance Se. The role of the impact parameter in the energy dissipation mechanism was demonstrated by the electronic stopping of Mg for high-energy protons. It was also shown that the impact parameter exerts a significant influence on the excitation of core electrons [36]. In addition, the measured SCS of Mg for protons and He ions is higher than that given by the FEG model. This disagreement was thought to be due to the overlap between the electronic orbitals of the He 1s and Mg 2p [37].
This paper presents a RT-TDDFT calculation of the Se of Mg for protons moving along both channeling and off-channeling trajectories. Extending beyond our previous investigations [31] in which the excitation effect of 2p electron of Mg is primarily studied. In the present paper, we further studied the excitation effect of 2s electron excitation of Mg by constructing new pseudopotentials with explicitly accounting for this inner electron configuration, investigated the threshold effect of the inner-electron excitation, and revealed the contribution of 2s electron excitation to the Se in the high-energy region. The results reported in this paper are crucial for a comprehensive understanding of the electronic stopping of Mg for protons.
2. Model and method
In the present study, a combined approach of RT-TDDFT model and non-adiabatic dynamics [38, 39] is utilized to investigate the Se of magnesium for protons. This method can be used to study the dynamics of excited electronic states and the energy transport between incident particles and electronic subsystem of solids. The time-dependent Kohn–Sham (KS) equation [38] is employed to describe the time evolution of the electrons (atomic units are used henceforth):
$\begin{eqnarray}{\rm{i}}\frac{\partial }{\partial t}{\varphi }_{j}({\boldsymbol{r}},t)=\left[-\frac{1}{2}{{\rm{\nabla }}}^{2}+{v}_{{\rm{KS}}}\left[n\right]({\boldsymbol{r}},t)\right]{\varphi }_{j}({\boldsymbol{r}},t),\end{eqnarray}$
where φj(r, t) is the jth time-dependent Kohn–Sham orbital, which depends on the spatial coordinates r and time t. ${v}_{{\rm{KS}}}\left[n\right]({\boldsymbol{r}},t)$ is the time-dependent Kohn–Sham effective potential, which is divided into the following terms: $\begin{eqnarray}{v}_{{\rm{KS}}}[n]({\boldsymbol{r}},t)={v}_{\,\rm{ext}\,}({\boldsymbol{r}},t)+{v}_{{\rm{H}}}[n]({\boldsymbol{r}},t)+{v}_{{\rm{XC}}}[n]({\boldsymbol{r}},t),\end{eqnarray}$
in which vext(r, t) is the time-dependent electron-nucleus potential, vH[n](r, t) is the Hartree potential for describing the classical Coulombic interactions among electrons, and the vXC[n](r, t) denotes the time-dependent exchange-correlation potential. The electron density n(r, t) of the system is given by $\begin{eqnarray}n({\boldsymbol{r}},t)=2\displaystyle \sum _{j=1}^{N/2}| {\varphi }_{j}({\boldsymbol{r}},t){| }^{2}.\end{eqnarray}$
In this work, the interaction between electrons and ionic cores is described by Optimized Norm-Conserving Vanderbilt (ONCV) pseudopotential [40]. Conventionally, only one electron state per angular momentum is taken into account by the norm-conserving pseudopotential, while two electron states for the same angular momentum can be considered by the ONCV pseudopotential. In this paper, three pseudopotentials are constructed for the Mg atom with different electron configurations: Mg2 ([Ne]3s2), Mg8 ([He2s2]2p63s2), Mg10 ([He]2s22p63s2). These pseudopotentials are employed to investigate the contribution of the inner 2s and 2p electron excitation to the Se. The Perdew–Burke–Ernzerhof (PBE) functional parametrization [41] is adopted for the exchange-correlation potential. It improves upon the local density approximation (LDA) by incorporating correction for the electron density gradient, leading to more accurate predictions of the electronic structure of Mg.
In this study, a 3 × 3 × 3 supercell containing 54 magnesium atoms is employed, periodic boundary conditions and Ewald summation are considered [42]. Additionally, a 4 × 4 × 4 supercell containing 128 magnesium atoms was constructed to investigate the effect of supercell size on the accuracy of Se calculations. A comparison was made between the Se derived from the 4 × 4 × 4 supercell and that obtained from the present 3 × 3 × 3 supercell for the midpoint trajectory at 4.0 au. The discrepancy in the Se obtained from the two supercells was found to be within 1% for all pseudopotentials. The experimental lattice parameters of hexagonal close packed (hcp) structured Mg are used, where a = b = 3.21 Å, c = 5.21 Å, α = β = 90∘, γ = 120∘. The wavefunction, electron density, and external potential are discretized in a real-space grid along all three spatial coordinates in the simulation cell with a uniform spacing of 0.16 Å. A single k-point (Γ) was used to integrate the Brillouin zone. We also calculated the Se by a 2 × 2 × 2 k-point Monkhorst–Packgrid, and the Se for selected velocities exhibits a relative error of less than 1%.
The electronic ground state of magnesium is obtained by diagonalizing the time-independent Kohn–Sham Hamiltonian. Subsequently, the projectile is placed on the upper boundary of the simulation box and an initial velocity is given to the projectile instantaneously at the beginning of the time evolution simulation. The host atoms are fixed at equilibrium positions during the real-time evolution [43, 44], which ensures that the projectile energy is transferred to the electronic subsystem of Mg through inelastic collisions. We set different propagation time steps for different velocities so that v × Δt ≈ 2.605 × 10−3 Å is kept, which ensures the conservation of the total energy of system. As the projectile moves along the channeling trajectory and off-channeling geometries, the kinetic energy of the projectile is gradually deposited into the electronic subsystem of magnesium through inelastic collisions with target atoms.
The projectile protons lose energy gradually when traversing through Mg, and the energy growth rate of the electronic subsystem of Mg is equal to the energy loss rate of protons. The instantaneous Se of Mg is defined as the loss rate of the projectile kinetic energy E(x) with respect to the projectile displacement x, Se = −dE(x)/dx. Figure 1 shows the kinetic energy loss of protons traversing along the midpoint trajectory at different velocities. It is worth noting that at the beginning of time evolution, the sudden motion of the projectile and the capture of electrons from the host atoms result in a ‘transient’ energy loss before the ion reaches the equilibrium charged state. This effect is particularly pronounced for low-velocity protons [44]. To determine the channeling Se for equilibrium-charged protons, non-equilibrium contributions are eliminated from the energy loss profile. The equilibrium Se is derived by averaging the instantaneous Se over two lattice periods between the two vertical dashed lines, as shown in Figure 1. It can be seen that after reaching the equilibrium charge state, the energy loss of the protons exhibits periodic oscillation that correlates with the lattice structure of bulk Mg.
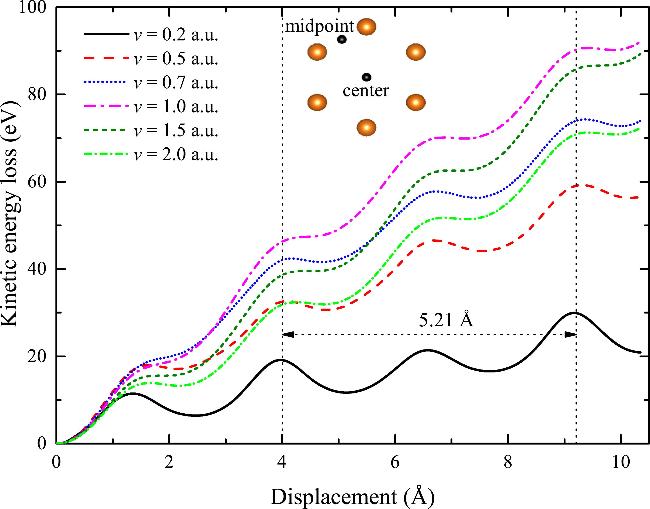
Figure 1. The kinetic energy loss of protons moving along the midpoint trajectory obtained from Mg10 pseudopotential model as a function of the displacement for different velocities. The inset shows the top view of the incidence geometry, the orange spheres indicate Mg atoms and the black circles signify projectile impact locations. |
In order to model the random collisions that occur in most experiments, the simulations in this study not only permit protons to move in a crystal axis direction, but also allow them to traverse the magnesium lattice in random directions (off-channeling trajectories). The projectiles occasionally interact with the tightly bound inner electrons of the host atoms in the off-channeling condition. In this study, 12 distinct off-channeling trajectories are randomly specified within the lattice. The smallest impact parameter for the off-channeling trajectories is 0.472 Å. However, these trajectories are selected by three guidelines [45]: (1) the head-on collision between protons and Mg atoms is avoided; (2) the secondary excitations induced by protons due to their repeated entrance into the simulation box is avoided; (3) as many collision parameters as possible are sampled by the chosen trajectories. The Se for a single off-channeling trajectory is computed by averaging instantaneous stopping. The off-channeling Se represents the mean value derived from 12 random trajectories sampled at given velocity. The TDDFT simulations are performed by the OCTOPUS computational package [46, 47].
3. Result and discussion
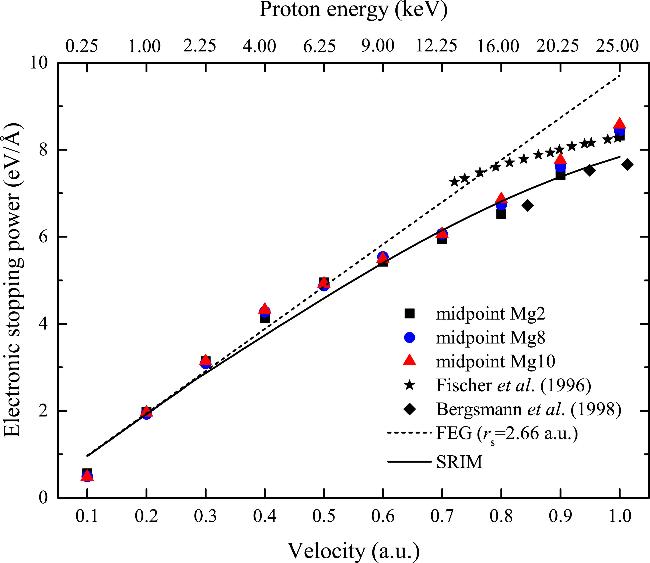
The Se of Mg for protons traveling along the midpoint trajectory in the low-energy range is shown in Figure 2. Additionally, the experimental stopping data [37, 48], FEG predictions [49] with Wigner-Seitz radius rs = 2.66 au (3s2) and SRIM package predictions [50] are also shown in the figure for comparison. The experimental data were obtained from off-channeling trajectories. The results of SRIM package are based on an extension of Lindhard–Scharff–Schiøtt (LSS) theory [51], with input from existing experimental data. The LSS theory provides a theoretical model for the energy loss of heavy ions in solids, which is capable of calculating the average energy loss rate of ions as they penetrate through materials. It can be seen from Figure 2 that, the Se calculated by TDDFT deviates from the velocity proportionality and aligns well with one set of experimental data [37] in the low-energy region. This can be attributed to the influence of the charge exchange effect between protons and host electrons. The Se obtained from Mg2, Mg8, and Mg10 pseudopotentials are almost identical in the low energy region. This suggests that the inner electrons of Mg are not excited in the midpoint trajectory when v < 1.0 au. Additionally, it indicates that the 3s electron is sufficient to describe the Se of Mg for low energy protons.
In order to study the threshold effect of inner-electron excitation of Mg, the density of electron states of Mg was calculated, as shown in Figure 3. The 3s band of magnesium extends up to the Fermi energy level, indicating the absence of a discernible threshold effect in the excitation of the 3s electrons [52, 53]. The occupied 2p state of Mg is located at 43 eV below the Fermi energy level, and the occupied 2s state of Mg is located at 76.18 eV below the Fermi level. In comparison to the experimental data of 49.6 and 88.6 eV [54], the relative errors of the calculated band offsets are 13.3% and 14%, respectively. In contrast, the relative errors of the band offsets obtained using the norm-conserving pseudopotential are 35.5% and 19.86% [36], respectively. This suggests that the ground state electronic structure of Mg is more accurately given by the ONCV pseudopotentials constructed in this study. This finding indicates that the equilibrium electronic structure of bulk Mg is well described by the ONCV pseudopotentials in this work, and this accurate ground state electronic structure is fundamental for predicting excited properties of electrons, including the electronic stopping power.
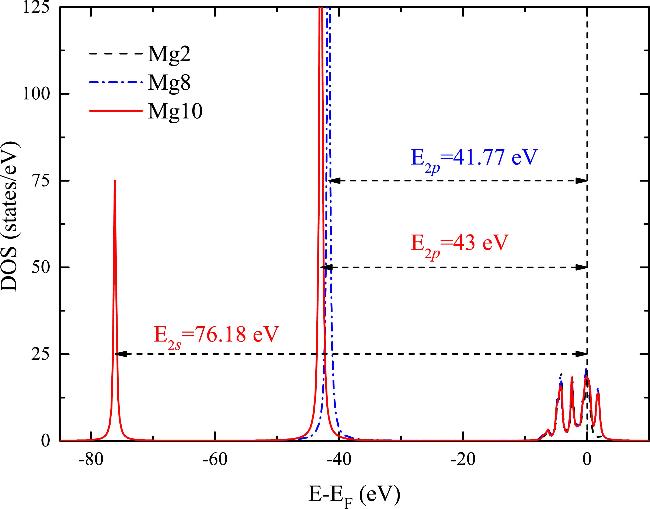
Figure 3. The density of electron states of Mg for the three pseudopotential models. The Fermi energy level is shifted to 0 eV and indicated by the dashed vertical line. |
The threshold velocity for excitation of 2p electrons to the Fermi energy level can be calculated from the following equation [44], 5 ), the threshold velocity for 2p and 2s electron excitation is calculated to be vth = 1.09 au and vth = 1.22 au, respectively. The results of the ground state calculations show that there is no inner-electron excitation at v < 1.09 au, which is consistent with the results of the TDDFT calculations, as shown in Figure 2. It indicates that the excitation of the 3s electron plays a dominant role in the low energy electronic stopping of magnesium.
$\begin{eqnarray}{v}_{{\rm{th}}}=\frac{{\rm{\Delta }}}{2\hslash {k}_{{\rm{F}}}},\end{eqnarray}$
where Δ denotes the band offset of the specific state, corresponding to the energy interval between the edge of the specific energy band and the Fermi energy level of Mg. The Fermi wave vector kF is calculated as 1.92/rs = 0.72/a0 for the FEG including only 3s electrons of Mg (rs = 2.66 au) and kF = 1.14/a0 for the effective uniform electron gas containing both 2p and 3s electrons of Mg (rs = 1.68 au). According to equation (The Se of protons is extended to higher velocities, as shown in Figure 4. In order to investigate the microscopic mechanism for inner electron excitation of Mg, the stopping power of proton moving in the center trajectory was calculated at two velocities (2.0 and 3.0 au). It is found that the difference in the Se obtained from the three pseudopotentials is negligible in the center trajectory, and the Se calculated from the center trajectory is consistent with the off-channeling Se obtained from the Mg2 pseudopotential, which indicates that there is no inner-electron excitation at this collision parameter. In contrast, for the midpoint trajectory with a small collision parameter, the Se of the Mg8 and Mg10 pseudopotentials is significantly higher and there are some discrepancies in the Se obtained from three Mg pseudopotentials for high energy protons, suggesting that the inner electron excitation is triggered under the midpoint trajectory. For the Mg2 pseudopotential, the off-channeling Se is slightly higher than that of the midpoint trajectory when v < 1.0 au, and a little lower than that of the midpoint trajectory between 1.0 and 3.0 au. The Se is almost the same for the two incident geometries when v > 3.0 au. Compared to the Se obtained for the midpoint trajectory, the off-channeling Se derived from Mg8 and Mg10 pseudopotentials is significantly enhanced over the velocity range considered. This suggests that the contribution of inner-electron excitation to Se depends on the impact parameter.
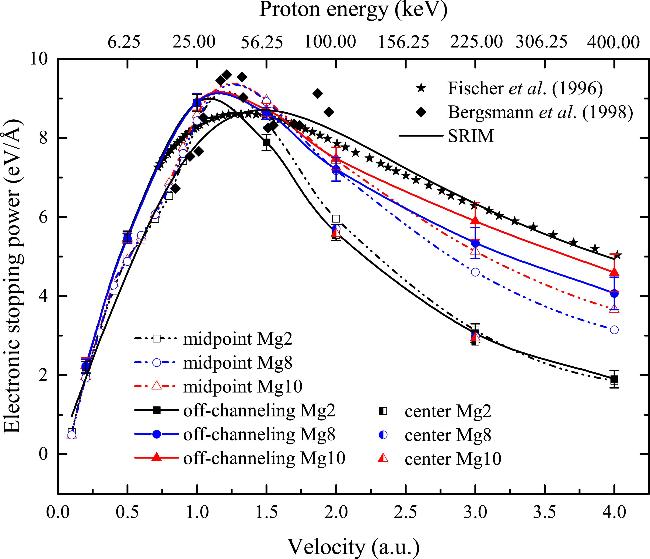
Figure 4. The channeling and off-channeling electronic stopping power of Mg as a function of proton velocity. The channeling stopping is calculated for protons moving along the midpoint trajectory and the center trajectory. Experimental data [37, 48] are also shown for comparison. The solid line represents the electronic stopping predicted by the SRIM package [50]. |
In the off-channeling geometry, the Se including 2p electron (Mg8 pseudopotential) deviates from that of the Mg2 pseudopotential at v > 1.0 au, and the discrepancy increases as proton velocity increases. The Se obtained from the Mg10 pseudopotential also starts to departure from that of the Mg8 pseudopotential when v > 1.5 au, and the discrepancy increases gradually with the velocity increasing. This suggests that the inner-electron excitation occurs mainly in the high-velocity range. It is observed that the threshold velocities for 2p and 2s electron excitation are 1.0 and 1.5 au by the TDDFT calculations, respectively. However, the threshold velocities for 2p electron excitation (1.09 au) and 2s electron excitation (1.22 au) calculated by equation (5 ) differ from those calculated by TDDFT simulations. The discrepancy may stem from equation (5 ) generally considers only the band offset of the target materials, while ignoring the properties of the projectile ions, such as the electron capture effect. Moreover, the discrepancy between the Se of the Mg10 pseudopotential and that of the Mg8 pseudopotential is less than that between Mg8 pseudopotential and Mg2 pseudopotential. This indicates that the contribution of 2s electron excitation to the Se is less than that of the 2p electron excitation. This can be attributed to the fact that the 2s subshell is located at a much lower energy level, with a smaller number of electrons compared to the 2p subshell.
As can be seen in Figure 4, the off-channeling Se including also 2s and 2p electrons agrees well with the experimental data [48] and the SRIM predictions in the velocity range beyond the Bragg peak, and is only 8.8% lower than the experimental stopping at the velocity of 4.0 au This indicates that the excitation of inner electrons is enhanced in the off-channeling geometry, and the excitation efficiency of the inner electrons is higher in comparison with that in the midpoint trajectory. This is due to the fact that the inner electrons are highly localized around the target nuclei, which means that the excitation of core electrons occurs in close collisions. The off-channeling geometry is conducive to the excitation of core electrons by sampling a wider range of collision parameters and allowing close proximity to target atoms.
To explore the microscopic mechanism responsible for the electronic stopping and ensure the accuracy of TDDFT predictions for the electronic stopping of solids, it is important to guarantee that the electronic structure of solids in ground state is well reproduced by the pseudopotential model employed. Because the current understanding is that the electronic energy loss of energetic protons in solids is closely related to the electronic structure of materials [44]. It is therefore essential to obtain an accurate ground-state electronic structure first of all so as to reasonably describe the properties of excited electronic states, including electronic energy loss. Furthermore, it is crucial for the pseudopotential to accurately capture the behavior of inner-electron excitation in close collisions. The pseudopotentials constructed in this study effectively describe both the ground-state electronic structure of magnesium and the excitation dynamics of inner electrons.
The number of electrons excited from different orbitals is extracted to display the electronic excitation in Mg more intuitively. The number of electronic excitations, nex(t), can be determined by projecting the time-dependent Kohn–Sham wavefunction ${\varphi }_{{n}^{{\prime} }}(t)$ onto the ground-state wavefunction φn, which is expressed as follows [55],
$\begin{eqnarray}{n}_{{\rm{ex}}}(t)=\displaystyle \sum _{n{n}^{{\prime} }}^{{n}_{{\rm{occ}}}}{f}_{n}\left[1-| \langle {\phi }_{n}| {\varphi }_{{n}^{{\prime} }}(t)\rangle {| }^{2}\right].\end{eqnarray}$
The nocc refers to the number of occupied states, while fn signifies the fixed occupation of the Kohn–Sham state φn (f = 2 for occupied states). The number of electrons excited from the Mg8 and Mg10 pseudopotentials was obtained for protons moving along different trajectories. The percentage of electron excited from each bands of Mg relative to the total number of electrons excited is shown in Figure 5. Figure 5(a) presents the ratio of the number of electrons excited obtained from the Mg8 pseudopotential, while Figure 5(b) illustrates the rate of electron excitation from the Mg10 pseudopotential. In order to explore the electron excitation rate at a large collision parameter, two velocities (2.0 and 3.0 au) were selected for the center trajectory.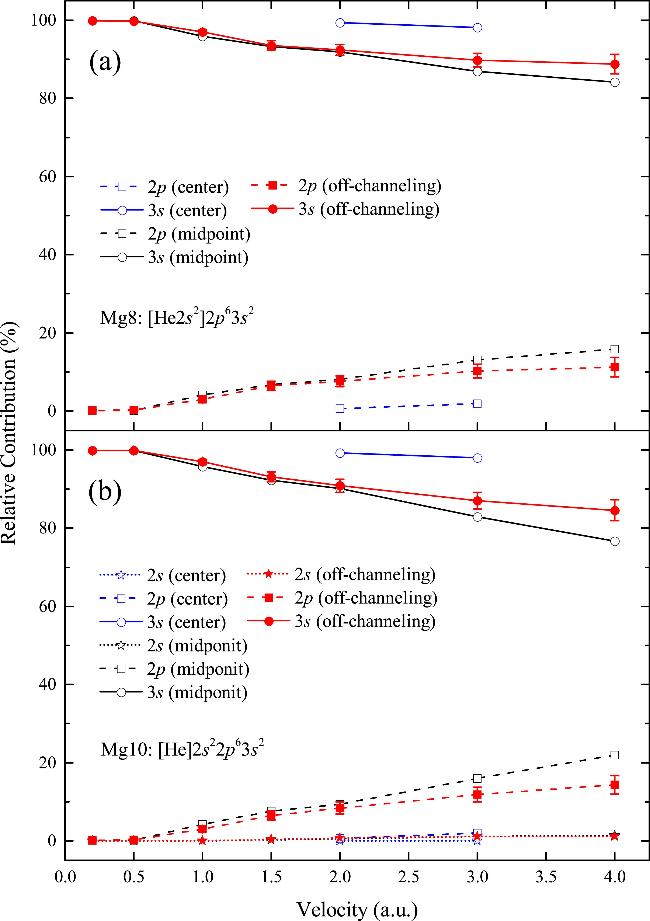
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. The relative contribution of electronic excitations of each electron band to the total number of electronic excitations obtained by (a) Mg8 and (b) Mg10 pseudopotentials, respectively. |
As displayed in Figure 5, the 3s electron excitation exhibits a major contribution throughout the velocity range, whether using the or Mg10 pseudopotential. On the other hand, the relative contribution of 2p electron excitation in Mg8 and Mg10 pseudopotentials increases with increasing velocity. For the midpoint trajectory, the percentages are as high as 15.87% and 21.93% at 4.0 au, respectively. In contrast, the relative contribution of the deeper 2s electrons is minimal, amounting to 1% at 4.0 au. This is due to the fact that the conduction electrons are located at higher energy levels and are susceptible to protons, which explains why conduction electron excitation accounts for the major contribution to the total electron excitations. In contrast, inner electrons residing at lower energy levels require greater energies to overcome the strong attraction with the atomic nucleus for excitation. In addition, inner electrons exhibit a strong shielding effect against external electric fields, which diminishes their susceptibility to external influences and makes their excitation less likely. Consequently, the excitation efficiency of inner electrons is relatively low.
Furthermore, the relative contribution of the 2p electrons in the midpoint trajectory exceeds that in the off-channeling trajectories, for both Mg8 and the Mg10 pseudopotentials. This is due to the fact that the total number of electrons excited in the off-channeling trajectories increases significantly compared to the midpoint trajectory, while the number of 2p electron excitations increases slightly. As a result, the proportion of 2p electron excitations in the total number of electron excitations is decreased in the off-channeling geometry in comparison with that in the midpoint trajectory. The results of the two velocity points for the center trajectory showed that the proportion of inner-electron excitations to the total electron excitations is significantly lower than those of other trajectories, suggesting that the relative contribution of the inner electrons is significantly diminished at the maximum impact parameter.
4. Conclusion
In this paper, the Se and electron excitation of Mg for protons are investigated based on RT-TDDFT combined with non-adiabatic dynamics simulations. This study examined the microscopic mechanism underlying the inner-electron excitation of Mg and its contribution to the electronic energy loss of protons by constructing new pseudopotential models of the Mg atom, which incorporate the inner electron configuration. It is found that the Se deviates from the linear relationship with velocity in the low-velocity range due to the charge exchange effect. The excitation of 3s electrons of magnesium dominates the low-energy Se. Compared to the Se that considers only conduction electron, the Se including also inner electron excitation is significantly enhanced in the high-energy region. Therefore, the inner-electron excitation plays a crucially important role in the high-energy Se of magnesium, and the inner-electron excitation of magnesium is crucially important to obtain an accurate Se in the high-energy region. The Se increases significantly at smaller collision parameters and is closer to the experimental data, suggesting that the inner-electron excitation is triggered by ion-electron direct collision.
From the point of view of the number of electrons excited in each shell, the valence electron excitation plays a major role in the whole velocity range considered in the present study. The fact that the 2s electrons are located at much lower energy levels leads to a low excitation efficiency for this inner electron shell. However, in terms of the contribution to Se, the 2p-electron excitation plays a more prominent role than the valence electron excitation in the high-velocity range. The Mg atomic pseudopotential models constructed in this study not only reproduce the electronic structure of the ground state of Mg well, but also provide a rational description of the excited state dynamics of the inner electrons under proton irradiation.